Differential Regulation of Telomeric Complex by BCR-ABL1 Kinase In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Introduction of Human Telomerase Reverse Transcriptase to Normal Human Fibroblasts Enhances DNA Repair Capacity
Vol. 10, 2551–2560, April 1, 2004 Clinical Cancer Research 2551 Introduction of Human Telomerase Reverse Transcriptase to Normal Human Fibroblasts Enhances DNA Repair Capacity Ki-Hyuk Shin,1 Mo K. Kang,1 Erica Dicterow,1 INTRODUCTION Ayako Kameta,1 Marcel A. Baluda,1 and Telomerase, which consists of the catalytic protein subunit, No-Hee Park1,2 human telomerase reverse transcriptase (hTERT), the RNA component of telomerase (hTR), and several associated pro- 1School of Dentistry and 2Jonsson Comprehensive Cancer Center, University of California, Los Angeles, California teins, has been primarily associated with maintaining the integ- rity of cellular DNA telomeres in normal cells (1, 2). Telomer- ase activity is correlated with the expression of hTERT, but not ABSTRACT with that of hTR (3, 4). Purpose: From numerous reports on proteins involved The involvement of DNA repair proteins in telomere main- in DNA repair and telomere maintenance that physically tenance has been well documented (5–8). In eukaryotic cells, associate with human telomerase reverse transcriptase nonhomologous end-joining requires a DNA ligase and the (hTERT), we inferred that hTERT/telomerase might play a DNA-activated protein kinase, which is recruited to the DNA role in DNA repair. We investigated this possibility in nor- ends by the DNA-binding protein Ku. Ku binds to hTERT mal human oral fibroblasts (NHOF) with and without ec- without the need for telomeric DNA or hTR (9), binds the topic expression of hTERT/telomerase. telomere repeat-binding proteins TRF1 (10) and TRF2 (11), and Experimental Design: To study the effect of hTERT/ is thought to regulate the access of telomerase to telomere DNA telomerase on DNA repair, we examined the mutation fre- ends (12, 13). -

Telomeres.Pdf
Telomeres Secondary article Elizabeth H Blackburn, University of California, San Francisco, California, USA Article Contents . Introduction Telomeres are specialized DNA–protein structures that occur at the ends of eukaryotic . The Replication Paradox chromosomes. A special ribonucleoprotein enzyme called telomerase is required for the . Structure of Telomeres synthesis and maintenance of telomeric DNA. Synthesis of Telomeric DNA by Telomerase . Functions of Telomeres Introduction . Telomere Homeostasis . Alternatives to Telomerase-generated Telomeric DNA Telomeres are the specialized chromosomal DNA–protein . Evolution of Telomeres and Telomerase structures that comprise the terminal regions of eukaryotic chromosomes. As discovered through studies of maize and somes. One critical part of this protective function is to fruitfly chromosomes in the 1930s, they are required to provide a means by which the linear chromosomal DNA protect and stabilize the genetic material carried by can be replicated completely, without the loss of terminal eukaryotic chromosomes. Telomeres are dynamic struc- DNA nucleotides from the 5’ end of each strand of this tures, with their terminal DNA being constantly built up DNA. This is necessary to prevent progressive loss of and degraded as dividing cells replicate their chromo- terminal DNA sequences in successive cycles of chromo- somes. One strand of the telomeric DNA is synthesized by somal replication. a specialized ribonucleoprotein reverse transcriptase called telomerase. Telomerase is required for both -

Expression of Telomerase Activity, Human Telomerase RNA, and Telomerase Reverse Transcriptase in Gastric Adenocarcinomas Jinyoung Yoo, M.D., Ph.D., Sonya Y
Expression of Telomerase Activity, Human Telomerase RNA, and Telomerase Reverse Transcriptase in Gastric Adenocarcinomas Jinyoung Yoo, M.D., Ph.D., Sonya Y. Park, Seok Jin Kang, M.D., Ph.D., Byung Kee Kim, M.D., Ph.D., Sang In Shim, M.D., Ph.D., Chang Suk Kang, M.D., Ph.D. Department of Pathology, St. Vincent’s Hospital, Catholic University, Suwon, South Korea esis of gastric cancer and may reflect, along with Telomerase is an RNA-dependent DNA polymerase enhanced hTR, the malignant potential of the tu- that synthesizes TTAGGG telomeric DNA onto chro- mor. It is noteworthy that methacarn-fixed tissue mosome ends to compensate for sequence loss dur- cannot as yet substitute for the frozen section in the ing DNA replication. It has been detected in 85–90% TRAP assay. of all primary human cancers, implicating that the telomerase seems to be reactivated in tumors and KEY WORDS: hTR, Stomach cancer, Telomerase, that such activity may play a role in the tumorigenic TERT. process. The purpose of this study was to evaluate Mod Pathol 2003;16(7):700–707 telomerase activity, human telomerase RNA (hTR), and telomerase reverse transcriptase (TERT) in Recent studies of stomach cancer have been di- stomach cancer and to determine their potential rected toward gaining a better understanding of relationships to clinicopathologic parameters. Fro- tumor biology. Molecular analysis has suggested zen and corresponding methacarn-fixed paraffin- that alterations in the structures and functions of embedded tissue samples were obtained from 51 oncogenes and tumor suppressor genes, genetic patients with gastric adenocarcinoma and analyzed instability, as well as the acquisition of cell immor- for telomerase activity by using a TRAPeze ELISA tality may be of relevance in the pathogenesis of kit. -

Fructose Causes Liver Damage, Polyploidy, and Dysplasia in the Setting of Short Telomeres and P53 Loss
H OH metabolites OH Article Fructose Causes Liver Damage, Polyploidy, and Dysplasia in the Setting of Short Telomeres and p53 Loss Christopher Chronowski 1, Viktor Akhanov 1, Doug Chan 2, Andre Catic 1,2 , Milton Finegold 3 and Ergün Sahin 1,4,* 1 Huffington Center on Aging, Baylor College of Medicine, Houston, TX 77030, USA; [email protected] (C.C.); [email protected] (V.A.); [email protected] (A.C.) 2 Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX 77030, USA; [email protected] 3 Department of Pathology, Baylor College of Medicine, Houston, TX 77030, USA; fi[email protected] 4 Department of Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030, USA * Correspondence: [email protected]; Tel.: +1-713-798-6685; Fax: +1-713-798-4146 Abstract: Studies in humans and model systems have established an important role of short telomeres in predisposing to liver fibrosis through pathways that are incompletely understood. Recent studies have shown that telomere dysfunction impairs cellular metabolism, but whether and how these metabolic alterations contribute to liver fibrosis is not well understood. Here, we investigated whether short telomeres change the hepatic response to metabolic stress induced by fructose, a sugar that is highly implicated in non-alcoholic fatty liver disease. We find that telomere shortening in telomerase knockout mice (TKO) imparts a pronounced susceptibility to fructose as reflected in the activation of p53, increased apoptosis, and senescence, despite lower hepatic fat accumulation in TKO mice compared to wild type mice with long telomeres. The decreased fat accumulation in TKO Citation: Chronowski, C.; Akhanov, is mediated by p53 and deletion of p53 normalizes hepatic fat content but also causes polyploidy, V.; Chan, D.; Catic, A.; Finegold, M.; Sahin, E. -
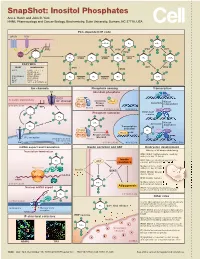
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

The Conserved Structure of Plant Telomerase RNA Provides the Missing Link for an Evolutionary Pathway from Ciliates to Humans
The conserved structure of plant telomerase RNA provides the missing link for an evolutionary pathway from ciliates to humans Jiarui Songa, Dhenugen Logeswaranb,1, Claudia Castillo-Gonzáleza,1, Yang Lib, Sreyashree Bosea, Behailu Birhanu Aklilua, Zeyang Mac,d, Alexander Polkhovskiya,e, Julian J.-L. Chenb,2, and Dorothy E. Shippena,2 aDepartment of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843; bSchool of Molecular Sciences, Arizona State University, Tempe, AZ 85287; cNational Maize Improvement Center of China, China Agricultural University, 100193 Beijing, China; dCollege of Agronomy and Biotechnology, China Agricultural University, 100193 Beijing, China; and eCenter of Life Sciences, Skolkovo Institute of Science and Technology, 121205 Moscow, Russian Federation Edited by Thomas R. Cech, University of Colorado Boulder, Boulder, CO, and approved October 24, 2019 (received for review September 4, 2019) Telomerase is essential for maintaining telomere integrity. Although transcribed by RNA polymerase III (Pol III) (6, 7). The La-related telomerase function is widely conserved, the integral telomerase protein P65 in Tetrahymena recognizes the 3′ poly-U tail of TR RNA (TR) that provides a template for telomeric DNA synthesis has and bends the RNA to facilitate telomerase RNP assembly (8, 9). diverged dramatically. Nevertheless, TR molecules retain 2 highly In contrast, fungi maintain much larger TR molecules (900 to conserved structural domains critical for catalysis: a template- 2,400 nt) that are transcribed by RNA polymerase II (Pol II) (3). proximal pseudoknot (PK) structure and a downstream stem-loop The 3′ end maturation of fungal TRs requires components of the structure. Here we introduce the authentic TR from the plant canonical snRNA biogenesis pathway and results in RNP assembly Arabidopsis thaliana, called AtTR, identified through next-generation sequencing of RNAs copurifying with Arabidopsis TERT. -

The Biochemical Activities of the Saccharomyces Cerevisiae Pif1 Helicase Are Regulated by Its N-Terminal Domain
G C A T T A C G G C A T genes Article The Biochemical Activities of the Saccharomyces cerevisiae Pif1 Helicase Are Regulated by Its N-Terminal Domain David G. Nickens y, Christopher W. Sausen y and Matthew L. Bochman * Molecular and Cellular Biochemistry Department, Indiana University, Bloomington, IN 47405, USA; [email protected] (D.G.N.); [email protected] (C.W.S.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 31 March 2019; Accepted: 20 May 2019; Published: 28 May 2019 Abstract: Pif1 family helicases represent a highly conserved class of enzymes involved in multiple aspects of genome maintenance. Many Pif1 helicases are multi-domain proteins, but the functions of their non-helicase domains are poorly understood. Here, we characterized how the N-terminal domain (NTD) of the Saccharomyces cerevisiae Pif1 helicase affects its functions both in vivo and in vitro. Removal of the Pif1 NTD alleviated the toxicity associated with Pif1 overexpression in yeast. Biochemically, the N-terminally truncated Pif1 (Pif1DN) retained in vitro DNA binding, DNA unwinding, and telomerase regulation activities, but these activities differed markedly from those displayed by full-length recombinant Pif1. However, Pif1DN was still able to synergize with the Hrq1 helicase to inhibit telomerase activity in vitro, similar to full-length Pif1. These data impact our understanding of Pif1 helicase evolution and the roles of these enzymes in the maintenance of genome integrity. Keywords: DNA helicase; Saccharomyces cerevisiae; Pif1; telomerase; telomere 1. Introduction DNA helicases are enzymes that couple DNA binding and ATP hydrolysis to unwind double-stranded DNA (dsDNA) into its component single strands [1]. -

DNA Bound by the Oxytricha Telomere Protein Is Accessible to Telomerase and Other DNA Polymerases DOROTHY E
Proc. Natl. Acad. Sci. USA Vol. 91, pp. 405-409, January 1994 Biochemistry DNA bound by the Oxytricha telomere protein is accessible to telomerase and other DNA polymerases DOROTHY E. SHIPPEN*, ELIZABETH H. BLACKBURNt, AND CAROLYN M. PRICE0§ tDepartment of Microbiology and Immunology, University of California, San Francisco, CA 94143; and tDepartment of Chemistry, University of Nebraska, Lincoln, NB 68588 Contributed by Elizabeth H. Blackburn, August 25, 1993 ABSTRACT Macronuclear telomeres in Oxytricha exist as oftelomere protein in these two populations is not altered by DNA-protein complexes in which the termini of the G-rich additional nuclease treatment. strands are bound by a 97-kDa telomere protein. During The fragment of DNA bound by the majority of telomere telome'ic DNA replication, the replication machinery must protein molecules corresponds to the most terminal 13 or 14 have access to the G-rich strand. However, given the stability nucleotides of the T4G4T4G4 overhang (4). Dimethyl sulfate of telomere protein binding, it has been unclear how this is footprinting demonstrated that the complex formed between accomplished. In this study we investigated the ability of the telomere protein and the residual DNA fragment retains several different DNA polymerases to access telomeric DNA in the same DNA-protein contacts present at native telomeres Oxytricha telomere protein-DNA complexes. Although DNA (4). Thus, these telomeric DNA-protein complexes are useful bound by the telomere protein is not degraded by micrococcal substrates for in vitro investigations of telomere structure nuclease or labeled by terminal deoxynucleotidyltrnsferase, (10). In this study we have employed the DNA-protein this DNA serves as an efficient primer for the addition of complexes to analyze the interaction of protein-bound telo- telomeric repeats by telomerase, a specialized RNA-dependent meric DNA with components of the DNA replication ma- DNA polymerase (ribonucleoprotein reverse tanscriptase), chinery. -

Telomere and Telomerase in Oncology
Cell Research (2002); 12(1):1-7 http://www.cell-research.com REVIEW Telomere and telomerase in oncology JIAO MU*, LI XIN WEI International Joint Cancer Institute, Second Military Medical University, Shanghai 200433, China ABSTRACT Shortening of the telomeric DNA at the chromosome ends is presumed to limit the lifespan of human cells and elicit a signal for the onset of cellular senescence. To continually proliferate across the senescent checkpoint, cells must restore and preserve telomere length. This can be achieved by telomerase, which has the reverse transcriptase activity. Telomerase activity is negative in human normal somatic cells but can be detected in most tumor cells. The enzyme is proposed to be an essential factor in cell immortalization and cancer progression. In this review we discuss the structure and function of telomere and telomerase and their roles in cell immortalization and oncogenesis. Simultaneously the experimental studies of telomerase assays for cancer detection and diagnosis are reviewed. Finally, we discuss the potential use of inhibitors of telomerase in anti-cancer therapy. Key words: Telomere, telomerase, cancer, telomerase assay, inhibitor. Telomere and cell replicative senescence base pairs of the end of telomeric DNA with each Telomeres, which are located at the end of round of DNA replication. Hence, the continual chromosome, are crucial to protect chromosome cycles of cell growth and division bring on progress- against degeneration, rearrangment and end to end ing telomere shortening[4]. Now it is clear that te- fusion[1]. Human telomeres are tandemly repeated lomere shortening is responsible for inducing the units of the hexanucleotide TTAGGG. The estimated senescent phenotype that results from repeated cell length of telomeric DNA varies from 2 to 20 kilo division, but the mechanism how a short telomere base pairs, depending on factors such as tissue type induces the senescence is still unknown. -

Commentary Tracking Telomerase
Cell, Vol. S116, S83–S86, January 23, 2004 Copyright 2004 by Cell Press Tracking Telomerase Commentary Carol W. Greider1 and Elizabeth H. Blackburn2,* neous size of the fragments in gel electrophoresis was 1Department of Molecular Biology and Genetics the first suggestion of unusual behavior of telomeric Johns Hopkins University School of Medicine DNA. A similar telomere repeat sequence, CCCCAAAA 725 North Wolfe Street was soon found on natural chromosome ends in other Baltimore, Maryland 21205 ciliates (Klobutcher et al., 1981). Another very unusual 2 Department of Biochemistry and Biophysics finding regarding these repeated sequences came in University of California, San Francisco 1982: David Prescott found that these repeated sequences Box 2200 are added de novo to ciliate chromosomes during the San Francisco, California 94143 developmental process of chromosome fragmentation (Boswell et al., 1982). This was the first hint that a special mechanism may exist to add telomere repeats. The Telomere Problem The next clue came from work in yeast. In a remarkable The paper reprinted here, the initial identification of telo- example of functional conservation across phylogenetic merase, resulted from our testing a very specific hypoth- kingdoms, Liz and Jack Szostak (Szostak and Black- esis: that an enzyme existed, then undiscovered, that burn, 1982) showed that the Tetrahymena telomeric se- could add telomeric repeats onto chromosome ends. quences could replace the yeast telomere entirely. A We based this hypothesis on several unexplained facts mini-chromosome with these foreign telomeres main- and creative questions being asked by people who were tained its linear structure and replicated and segregated trying to understand those facts. -

Purification of Tetrahymena Telomerase and Cloning of Genes Encoding the Two Protein Components of the Enzyme
Cell, Vol. 81,677-686, June 2, 1995, Copyright © 1995 by Cell Press Purification of Tetrahymena Telomerase and Cloning of Genes Encoding the Two Protein Components of the Enzyme Kathleen Collins, Ryuji Kobayashi, RNA component contains a 9 nt sequence (3'-AACCCC- and Carol W. Greider AAC-5~ complementary to the 6 nt telomeric repeat, Cold Spring Harbor Laboratory d(TTGGGG) (Greider and Blackburn, 1989). This region Cold Spring Harbor, New York 11724 of the telomerase RNA templates the synthesis of telo- meres in vivo (Yu et al., 1990). Additional telomerase RNAs have been cloned from both ciliates and yeast (Shippen- Summary Lentz and Blackburn, 1990; Romero and Blackburn, 1991 ; Lingner et al., 1994; Melek et al., 1994; Singer and Gott- Telomerase is a ribonucleoprotein DNA polymerase schling, 1994). Aside from the presence of a template re- that catalyzes the de novo synthesis of telomeric sim- gion, these RNAs lack extensive primary sequence con- ple sequence repeats. We describe the purification of servation. Phylogenetic evidence suggests a conserved telomerase and the cloning of cDNAs encoding two secondary structure in the ciliate RNAs (Lingner et al., protein subunits from the ciliate Tetrahymena. Two 1994; Romero and Blackburn, 1991), but the function of proteins of 80 and 95 kDa copurified and coimmuno- the conserved structures is not yet known. precipitated with telomerase activity and the pre- Models for primer recognition by Tetrahymena telo- viously identified Tetrahymena telomerase RNA. The merase have proposed two primer-binding sites to account p95 subunit specifically cross-linked to a radiolabeied for the differences in elongation processivity and binding telomeric DNA primer, while the p80 subunit specifi- affinity of different primers (see, for example, Collins and cally bound to radiolabeled telomerase RNA. -

Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt
International Journal of Molecular Sciences Article Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt 1,2, 1, 2 2 Stephanie Schwalm y, Martin Erhardt y, Isolde Römer , Josef Pfeilschifter , Uwe Zangemeister-Wittke 1,* and Andrea Huwiler 1,* 1 Institute of Pharmacology, University of Bern, Inselspital, INO-F, CH-3010 Bern, Switzerland; [email protected] (S.S.); [email protected] (M.E.) 2 Institute of General Pharmacology and Toxicology, University Hospital Frankfurt am Main, Goethe-University, Theodor-Stern Kai 7, D-60590 Frankfurt am Main, Germany; [email protected] (I.R.); [email protected] (J.P.) * Correspondence: [email protected] (U.Z.-W.); [email protected] (A.H.); Tel.: +41-31-6323214 (A.H.) These authors contributed equally to this work. y Received: 29 November 2019; Accepted: 12 February 2020; Published: 19 February 2020 Abstract: Ceramide kinase (CerK) is a lipid kinase that converts the proapoptotic ceramide to ceramide 1-phosphate, which has been proposed to have pro-malignant properties and regulate cell responses such as proliferation, migration, and inflammation. We used the parental human breast cancer cell line MDA-MB-231 and two single cell progenies derived from lung and bone metastasis upon injection of the parental cells into immuno-deficient mice. The lung and the bone metastatic cell lines showed a marked upregulation of CerK mRNA and activity when compared to the parental cell line. The metastatic cells also had increased migratory and invasive activity, which was dose-dependently reduced by the selective CerK inhibitor NVP-231.