Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase†,‡ Kap Lim,§ Randy J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 4. Enzyme Kinetics
Chapter 4. Enzyme Kinetics Enzyme Engineering 4.1 Why is enzyme kinetics important? 1. It provides valuable information for enzyme mechanism 2. It gives an insight into the role of an enzyme under physiological conditions 3. It can help show how the enzyme activity is controlled and regulated 1 4.2 How to obtain enzyme kinetics? Measure the formation of product or disappearance of substrate Mg2+ D-Glucose + ATP D-Glucose-6-phosphate + ADP 1. Discontinuous method Add the enzyme, then stop the reaction at different time by adding acid to the sample Measure the concentration of substrate or product using HPLC or other chromatography 4.2 How to obtain enzyme kinetics? 2. Continuous method Mg2+ D-Glucose + ATP D-Glucose-6-phosphate + ADP NADP+ NADPH D-Glucono-δ-lactone 6-phosphate + ADP NADPH absorb at 340nm, while NADP+ does not If a coupled reaction is used, the second reaction should not be rate-limiting (sufficient amount of the enzyme must be provided) Continuous method is not always available 2 4.2 How to obtain enzyme kinetics? Radioactive or fluorescent-labeling is useful Precautions The substrate, buffers, etc. must be extremely pure Enzyme should be pure Enzyme should be stable as the assay is proceeded * Temp, pH must be maintained carefully Once steady state is achieved, reaction rate must be constant and proportional to the enzyme added Initial rate of reaction should be measured to avoid possible complexity 4.3 How to analyze kinetic data? 4.3.1 One substrate reactions k 1 k2 E + S ES E + P k-1 Equilibrium assumption (1913) Second reaction is slower than first reverse reaction (k-1 >> k2) Vmax[S] V0 = Michaelis-Menten eqn K s +[S] [E][S] Vmax = k2[E]0 K = s [ES] 3 4.3 How to analyze kinetic data? Steady state assumption (1925) [ES] is constant Vmax[S] V0 = Km +[S] Vmax = k2[E]0 k2 + k−1 Km = k1 4.3.1 One substrate reactions Lineweaver-Burk equation 1 K 1 1 = m + v [S] Vmax Vmax Eddie-Hofstee equation v V v = max − [S] Km Km 4 4.3.1 One substrate reactions Significance of the result 1. -

Phosphotransferase Activity of Liver Mitochondria (Oxidative Phosphorylation/Adenine Nucleotide Ni-Oxides/Substrate Specificity/In Vivo Phosphorylation) G
Proc. Nat. Acad. Sci. USA Vol. 71, No. 11, pp. 4630-4634, November 1974 Participation of N1-Oxide Derivatives of Adenine Nucleotides in the Phosphotransferase Activity of Liver Mitochondria (oxidative phosphorylation/adenine nucleotide Ni-oxides/substrate specificity/in vivo phosphorylation) G. JEBELEANU, N. G. TY, H. H. MANTSCH*, 0. BARZU, G. NIACt, AND I. ABRUDAN Department of Biochemistry, Medical and Pharmaceutical Institute, Cluj, * Institute of Chemistry, University of Cluj, Cluj, and t Department of Physical Chemistry, University of Craiova, Craiova, Romania Communicated by Henry Lardy, September 3, 1974 ABSTRACT The modified adenine nucleotides ATP- rivatives of adenine nucleotides once produced become "ac- NO, ADP-NO, and AMP-NO were tested as potential tive" components of the mitochondrial or cellular adenylate substrates and/or inhibitors of mitochondrial phospho- transferases. ADP-NO is not recognized by the translocase pool. membrane; system located in the inner mitochondrial AND METHODS however, it is rapidly phosphorylated to ATP-NO in the MATERIALS outer compartment of mitochondria, by way of the The following commercially available chemicals were used: nucleosidediplhosphate kinase (EC 2.7.4.6) reaction, pro-' de- vitded there is sufficient ATP in the mitochondria. AMP- crystalline bovine serum albumin, glucose-6-phosphate NO is not phosphorylated by liver mitochondria to the hydrogenase (Glc-6-P dehydrogenase; EC 1.1.1.49; BDH corresponding nucleoside diphosphate; it cannot serve as Chemicals, Ltd.), yeast hexokinase (EC 2.7.1.1) (Nutritional substrate for adenylate kinase (EC 2.7.4.3). ATP-NO and Biochemicals, Cleveland), (log muscle lactate dehydrogenase ADP-NO, however, are substrates of this enzyme. -

Isozymes of Pyruvate Kinase in Liver and Hepatomas of the Rat1
[CANCER RESEARCH 34, 1439-1446, June 1974] Isozymes of Pyruvate Kinase in Liver and Hepatomas of the Rat1 Francis A. Farina,2 Jennie B. Shatton, Harold P. Morris, and Sidney Weinhouse The Fels Research Institute and the Department of Biochemistry, Temple University School oj Medicine, Philadelphia. Pennsylvania IV140 (F. A. F., J. B. S.. S. W.\, and the Department of Biochemistry. Howard University School of Medicine. Washington. D. C. 20001 [H. P. M .\ SUMMARY 23, 32, 41, 57). These alterations involve the replacement of those isozymes that are under dietary and hormonal control Pyruvate kinase (PK) (EC 2.7.1.40) isozymes were assayed by the host, and that have important metabolic functions in in normal rat liver and a series of transplantable rat the adult differentiated liver by other isozymes which are hepatomas ranging widely in growth rate and degree of normally either low in, or absent from, the adult tissue. As differentiation, with the use of gradient elution by chloride part of an ongoing study of this phenomenon, we have ion from columns of DEAE-cellulose. In agreement with examined the alteration of PK3 (EC 2.7.1.40) isozymes in other studies, three noninterconvertible forms were found in the Morris hepatomas (30. 31), a series of chemically rat tissues: isozyme I, the major form in adult rat liver: induced, transplantable rat hepatomas ranging widely in isozyme II, the sole form in heart and skeletal muscle: and growth rate and degree of differentiation. This enzyme isozyme III, the sole form in poorly differentiated hepato occupies a key position in the metabolism of cells and, as we mas, the major form in normal kidney and lung, and the pointed out previously (27, 28. -

Catalase Kinetics
Experiment #4: Catalase Kinetics MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry 5.310 Laboratory Chemistry 1 EXPERIMENT #4 A Study of the Kinetics of the Enzyme Catalase and its Reaction 2 3 With H2O2. A Further Study on Protein Assay Quantitation of Catalase I. OVERVIEW OF THE EXPERIMENT In this experiment, the student will investigate the enzyme activity of catalase by studying the decomposition of H2O2 to form water and oxygen. Using an oxygen based pressure sensor the student measures the amount of oxygen produced and then calculates the rate of the enzyme catalyzed reaction under various conditions. The student also completes a Protein Assay on an unknown sample of catalase using the Coomassie® Plus Protein Assay from Pierce to determine the protein concentration of the sample. The correlation between the actual and calculated concentration gives an indication of the experimental skills used in carrying out the experiment. II. OBJECTIVES This is an integrated experiment that includes topics from physical chemistry and biochemistry. It is designed to introduce students to the basics of: • Studying the effects of reaction environment, including temperature, and varying substrate concentration on the rate of an enzyme-catalyzed reaction. • Combining physical chemistry and biochemistry, principles and theory, with the goal of determining various biochemical and biophysical constants for the enzyme catalase. • How to acquire experimental kinetic data for an enzyme catalyzed reaction. • Learning how to perform data manipulation in order to extract out information such as rate constants from experimental kinetic data. • Correct handling of UV-VIS Spectroscopy 1 This experiment was synthesized by John J. -

Estimation of the Overall Kinetic Parameters of Enzyme Inactivation Using an Isoconversional Method D
Estimation of the overall kinetic parameters of enzyme inactivation using an isoconversional method D. Oancea, Alexandrina Stuparu, Madalina Nita, Mihaela Puiu, Adina Raducan To cite this version: D. Oancea, Alexandrina Stuparu, Madalina Nita, Mihaela Puiu, Adina Raducan. Estimation of the overall kinetic parameters of enzyme inactivation using an isoconversional method. Biophysical Chemistry, Elsevier, 2008, 138 (1-2), pp.50. 10.1016/j.bpc.2008.09.003. hal-00501718 HAL Id: hal-00501718 https://hal.archives-ouvertes.fr/hal-00501718 Submitted on 12 Jul 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÔØ ÅÒÙ×Ö ÔØ Estimation of the overall kinetic parameters of enzyme inactivation using an isoconversional method D. Oancea, Alexandrina Stuparu, Madalina Nita, Mihaela Puiu, Adina Raducan PII: S0301-4622(08)00176-2 DOI: doi: 10.1016/j.bpc.2008.09.003 Reference: BIOCHE 5155 To appear in: Biophysical Chemistry Received date: 17 August 2008 Revised date: 2 September 2008 Accepted date: 3 September 2008 Please cite this article as: D. Oancea, Alexandrina Stuparu, Madalina Nita, Mi- haela Puiu, Adina Raducan, Estimation of the overall kinetic parameters of en- zyme inactivation using an isoconversional method, Biophysical Chemistry (2008), doi: 10.1016/j.bpc.2008.09.003 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Pyruvate Kinase: Function, Regulation and Role in Cancer
Pyruvate kinase: Function, regulation and role in cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Israelsen, William J., and Matthew G. Vander Heiden. “Pyruvate Kinase: Function, Regulation and Role in Cancer.” Seminars in Cell & Developmental Biology 43 (2015): 43–51. As Published http://dx.doi.org/10.1016/j.semcdb.2015.08.004 Publisher Elsevier Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/105833 Terms of Use Creative Commons Attribution-NonCommercial-NoDerivs License Detailed Terms http://creativecommons.org/licenses/by-nc-nd/4.0/ HHS Public Access Author manuscript Author Manuscript Author ManuscriptSemin Cell Author Manuscript Dev Biol. Author Author Manuscript manuscript; available in PMC 2016 August 13. Published in final edited form as: Semin Cell Dev Biol. 2015 July ; 43: 43–51. doi:10.1016/j.semcdb.2015.08.004. Pyruvate kinase: function, regulation and role in cancer William J. Israelsena,1,* and Matthew G. Vander Heidena,b,* aKoch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA bDepartment of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02115, USA Abstract Pyruvate kinase is an enzyme that catalyzes the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP in glycolysis and plays a role in regulating cell metabolism. There are four mammalian pyruvate kinase isoforms with unique tissue expression patterns and regulatory properties. The M2 isoform of pyruvate kinase (PKM2) supports anabolic metabolism and is expressed both in cancer and normal tissue. The enzymatic activity of PKM2 is allosterically regulated by both intracellular signaling pathways and metabolites; PKM2 thus integrates signaling and metabolic inputs to modulate glucose metabolism according to the needs of the cell. -

Determination of Enzyme Kinetics in the Eppendorf Biospectrometer® Kinetic Using a Hexokinase and Glucose-6- Phosphate-Dehydrogenase from Saccharomyces Cerevisiae
USERGUIDE No. 39 I Detection I September 2014 Determination of enzyme kinetics in the Eppendorf BioSpectrometer® kinetic using a hexokinase and glucose-6- phosphate-dehydrogenase from Saccharomyces cerevisiae Martin Armbrecht, Eppendorf AG, Hamburg, Germany Abstract This Userguide will demonstrate measurement of enzyme activity using the Eppendorf BioSpectrometer. In order to optimize the measurement process, preliminary measurements were performed using the method “single continuous”. The actual activity measurements were then performed based on these results. Enzyme activities were determined via linear regression. For the measurements, a coupled reaction of a hexokinase and a glucose-6-phosphate-dehydrogenase from the baker’s yeast ‚ was used. Since the Eppendorf BioSpectrometer kinetic is equipped with a temperature controlled cuvette chamber, temperature dependence of this reaction could also be demonstrated. The highest activity was detected at 37 °C. Introduction Metabolic significance of the hexokinase reaction Activation of glucose via the hexokinase reaction Glycolysis Glucose degradation thus sustains respiration of all living beings. One central area within the metabolism of nearly all living beings is Prior to degradation, glucose needs to be rendered susceptible for the enzymatic degradation of glucose in the cytosol, or cytoplasma, degradation, i.e., activated. This process involves phosphorylation by respectively, in their cells. This process is called glycolysis. Glucose a hexokinase; with the expenditure of ATP, glucose-6-phosphate is is converted to 2 molecules of pyruvate in a stepwise fashion. One produced. Thus, the latter is the actual initial substrate of glycolysis molecule of glucose yields 2 ATP and 2 redox equivalents in the form (fig.1). In addition, glucose-6-phosphate is the initial substrate of a further of NADPH2. -
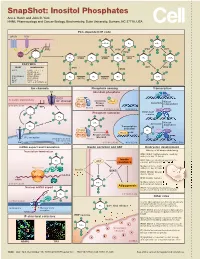
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

Effect of Enzyme/Substrate Ratio on the Antioxidant Properties of Hydrolysed African Yam Bean
African Journal of Biotechnology Vol. 11(50), pp. 11086-11091, 21 June, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB11.2271 ISSN 1684–5315 ©2012 Academic Journals Full Length Research Paper Effect of enzyme/substrate ratio on the antioxidant properties of hydrolysed African yam bean Fasasi Olufunmilayo*, Oyebode Esther and Fagbamila Oluwatoyin Department of Food Science and Technology, P. M. B. 704, Federal University of Technology, Akure, Nigeria. Accepted 8 June, 2012 The use of natural antioxidant as compared with synthetic antioxidant in food processing is a growing trend as consumers prefer natural to synthetic antioxidant mainly on emotional ground. This study investigates the antioxidant activity of hydrolysed African yam bean (Sphenostylis sternocarpa) which is regarded as one of the neglected underutilized species (NUS) of crop in Africa and Nigeria especially to improve food security and boost the economic importance of the crop. The antioxidant properties of African yam bean hydrolysates (AYH) produced at different enzyme to substrate (E/S) ratios of 1: 100 and 3: 100 (W/V) using pepsin (pH 2.0, 37°C) were studied. 2, 2-Diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity of the hydrolysates was significantly influenced by the E\S ratio as DPPH radical scavenging activity ranged from 56.1 to 75.8% in AYH (1: 100) and 33.3 to 58.8% in AYH (3:100) with 1000 µg/ml having the highest DPPH radical scavenging activity. AYH (1:100) and AYH (3:100) had higher reducing activities of 0.42 and 0.23, respectively at a concentration of 1000 µg/ml. -

Regulation of Energy Substrate Metabolism in Endurance Exercise
International Journal of Environmental Research and Public Health Review Regulation of Energy Substrate Metabolism in Endurance Exercise Abdullah F. Alghannam 1,* , Mazen M. Ghaith 2 and Maha H. Alhussain 3 1 Lifestyle and Health Research Center, Health Sciences Research Center, Princess Nourah bInt. Abdulrahman University, Riyadh 84428, Saudi Arabia 2 Faculty of Applied Medical Sciences, Laboratory Medicine Department, Umm Al-Qura University, Al Abdeyah, Makkah 7607, Saudi Arabia; [email protected] 3 Department of Food Science and Nutrition, College of Food and Agriculture Sciences, King Saud University, Riyadh 11451, Saudi Arabia; [email protected] * Correspondence: [email protected] Abstract: The human body requires energy to function. Adenosine triphosphate (ATP) is the cellular currency for energy-requiring processes including mechanical work (i.e., exercise). ATP used by the cells is ultimately derived from the catabolism of energy substrate molecules—carbohydrates, fat, and protein. In prolonged moderate to high-intensity exercise, there is a delicate interplay between carbohydrate and fat metabolism, and this bioenergetic process is tightly regulated by numerous physiological, nutritional, and environmental factors such as exercise intensity and du- ration, body mass and feeding state. Carbohydrate metabolism is of critical importance during prolonged endurance-type exercise, reflecting the physiological need to regulate glucose homeostasis, assuring optimal glycogen storage, proper muscle fuelling, and delaying the onset of fatigue. Fat metabolism represents a sustainable source of energy to meet energy demands and preserve the ‘limited’ carbohydrate stores. Coordinated neural, hormonal and circulatory events occur during prolonged endurance-type exercise, facilitating the delivery of fatty acids from adipose tissue to the Citation: Alghannam, A.F.; Ghaith, working muscle for oxidation. -

Prevalence of Pyruvate Kinase Deficiency
P3513 Prevalence of Red Cell Pyruvate Kinase Deficiency: A Systematic Literature Review Mike Storm1, Matthew H Secrest2*, Courtney Carrington2, Deb Casso2, Keely Gilroy1, Leanne Pladson1, Audra N Boscoe1 1Agios Pharmaceuticals Inc., Cambridge, MA, USA; 2IQVIA Epidemiology & Drug Safety, Seattle, WA and Cambridge, MA, USA; *Affiliation at the time research was conducted BACKGROUND METHODS (continued) RESULTS (continued) RESULTS (continued) • Pyruvate Kinase (PK) deficiency is a rare congenital hemolytic anemia Exclusion Criteria The remaining 34 studies were grouped based on methods and study Among these 4 studies, an important distinction was made between studies characterized by diminished activity of the PK enzyme in red blood population (Table 1). reporting diagnosed prevalence (n=3) and overall disease prevalence cells (RBC).1 • Non-human studies; (diagnosed and undiagnosed PK deficiency; n=1). Table 1. Distribution of extracted studies by type of study (n=34) • Low PK enzyme activity can lead to lifelong chronic hemolysis with • Publications that were not the primary report of the data • Two studies estimated diagnosed PK deficiency prevalence as 3.2 per associated symptoms and complications such as anemia, jaundice, (e.g., literature reviews); Type of study Number million4 and 8.5 per million5 by identifying diagnosed PK deficiency cases of studies gallstones, splenectomy and associated thrombosis, iron overload, and • Studies of PK deficiency prevalence/incidence conducted within a source from source populations of known size. liver cirrhosis.2 Population-based prevalence 2 population of patients with symptoms of PK deficiency such as anemia • We estimated the prevalence of diagnosed PK deficiency in a general • PK deficiency is caused by compound heterozygosity or homozygosity for or jaundice; Molecular PKLR screening in a general population 5 population to be 6.5 per million6 using data from another high-quality study 3 one or more of the >300 known mutations to the PKLR gene. -

Table S1. List of Oligonucleotide Primers Used
Table S1. List of oligonucleotide primers used. Cla4 LF-5' GTAGGATCCGCTCTGTCAAGCCTCCGACC M629Arev CCTCCCTCCATGTACTCcgcGATGACCCAgAGCTCGTTG M629Afwd CAACGAGCTcTGGGTCATCgcgGAGTACATGGAGGGAGG LF-3' GTAGGCCATCTAGGCCGCAATCTCGTCAAGTAAAGTCG RF-5' GTAGGCCTGAGTGGCCCGAGATTGCAACGTGTAACC RF-3' GTAGGATCCCGTACGCTGCGATCGCTTGC Ukc1 LF-5' GCAATATTATGTCTACTTTGAGCG M398Arev CCGCCGGGCAAgAAtTCcgcGAGAAGGTACAGATACGc M398Afwd gCGTATCTGTACCTTCTCgcgGAaTTcTTGCCCGGCGG LF-3' GAGGCCATCTAGGCCATTTACGATGGCAGACAAAGG RF-5' GTGGCCTGAGTGGCCATTGGTTTGGGCGAATGGC RF-3' GCAATATTCGTACGTCAACAGCGCG Nrc2 LF-5' GCAATATTTCGAAAAGGGTCGTTCC M454Grev GCCACCCATGCAGTAcTCgccGCAGAGGTAGAGGTAATC M454Gfwd GATTACCTCTACCTCTGCggcGAgTACTGCATGGGTGGC LF-3' GAGGCCATCTAGGCCGACGAGTGAAGCTTTCGAGCG RF-5' GAGGCCTGAGTGGCCTAAGCATCTTGGCTTCTGC RF-3' GCAATATTCGGTCAACGCTTTTCAGATACC Ipl1 LF-5' GTCAATATTCTACTTTGTGAAGACGCTGC M629Arev GCTCCCCACGACCAGCgAATTCGATagcGAGGAAGACTCGGCCCTCATC M629Afwd GATGAGGGCCGAGTCTTCCTCgctATCGAATTcGCTGGTCGTGGGGAGC LF-3' TGAGGCCATCTAGGCCGGTGCCTTAGATTCCGTATAGC RF-5' CATGGCCTGAGTGGCCGATTCTTCTTCTGTCATCGAC RF-3' GACAATATTGCTGACCTTGTCTACTTGG Ire1 LF-5' GCAATATTAAAGCACAACTCAACGC D1014Arev CCGTAGCCAAGCACCTCGgCCGAtATcGTGAGCGAAG D1014Afwd CTTCGCTCACgATaTCGGcCGAGGTGCTTGGCTACGG LF-3' GAGGCCATCTAGGCCAACTGGGCAAAGGAGATGGA RF-5' GAGGCCTGAGTGGCCGTGCGCCTGTGTATCTCTTTG RF-3' GCAATATTGGCCATCTGAGGGCTGAC Kin28 LF-5' GACAATATTCATCTTTCACCCTTCCAAAG L94Arev TGATGAGTGCTTCTAGATTGGTGTCggcGAAcTCgAGCACCAGGTTG L94Afwd CAACCTGGTGCTcGAgTTCgccGACACCAATCTAGAAGCACTCATCA LF-3' TGAGGCCATCTAGGCCCACAGAGATCCGCTTTAATGC RF-5' CATGGCCTGAGTGGCCAGGGCTAGTACGACCTCG