Hematology Assignment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

© 2019 First Aid for the USMLE Step 1 732 INDEX INDEX
Index A Abnormal uterine bleeding (AUB), heart failure, 306 Achondroplasia, 454 A-a gradient 618, 619 hypertension, 312 chromosome disorder, 64 in elderly, 654 adenomyosis, 634 naming convention for, 253 endochondral ossification in, 450 with hypoxemia, 654, 655 anemia with, 410 preload/afterload effects, 282 inheritance, 60 restrictive lung disease, 661 Asherman syndrome, 634 teratogenicity, 600 AChR (acetylcholine receptor), 229 Abacavir, 201, 203 leiomyoma (fibroid), 634 Acetaldehyde, 72 Acid-base physiology, 580 Abciximab, 122 polyps (endometrial), 634 Acetaldehyde dehydrogenase, 70, 72 Acidemia, 580 Glycoprotein IIb/IIIa inhibitors, thecoma, 632 Acetaminophen, 474 diuretic effect on, 595 429 ABO blood classification, 397 vs. aspirin for pediatric patients, 474 Acid-fast oocysts, 177 thrombogenesis and, 403 newborn hemolysis, 397 free radical injury and, 210 Acid-fast organisms, 125, 140, 155 Abdominal aorta, 357 Abruptio placentae, 626 hepatic necrosis from, 249 Acidic amino acids, 81 atherosclerosis in, 300, 687 cocaine use, 600 N-acetylcysteine for overdose, 671 Acid maltase, 86 bifurcation of, 649 preeclampsia, 629 for osteoarthritis, 458 Acidosis, 578, 580 Abdominal aortic aneurysm, 300 Abscess, 470 toxicity effects, 474 contractility in, 282 Abdominal colic acute inflammation and, 215 toxicity treatment for, 247 hyperkalemia with, 578 lead poisoning, 411 lung, 670 Acetazolamide, 252, 539, 594 Acid phosphatase in neutrophils, 398 Abdominal pain Absence seizures idiopathic intracranial Acid reflux bacterial peritonitis, 384 characteristics -

Erythrocytes: Overview, Morphology, Quantity by AH Rebar Et
In: A Guide to Hematology in Dogs and Cats, Rebar A.H., MacWilliams P.S., Feldman B.F., Metzger F.L., Pollock R.V.H. and Roche J. (Eds.). Publisher: Teton NewMedia, Jackson WY (www.veterinarywire.com). Internet Publisher: International Veterinary Information Service, Ithaca NY (www.ivis.org), 8-Feb-2005; A3304.0205 Erythrocytes: Overview, Morphology, Quantity A.H. Rebar1, P.S. MacWilliams2, B.F. Feldman 3, F.L. Metzger 4, R.V.H. Pollock 5 and J. Roche 6 1Dept of Veterinary Pathobiology, School of Veterinary Medicine, Purdue University, IN,USA. 2Dept of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, WI, USA. 3Dept of Biomedical Sciences & Pathobiology, VA-MD - Regional College of Veterinary Medicine, Virginia Tech, VA, USA. 4Metzger Animal Hospital,State College,PA, USA. 5Fort Hill Company, Montchanin, DE, USA. 6 Hematology Systems, IDEXX Laboratories, Westbrook, ME, USA. Overview Production Red blood cells (RBC) are produced in the bone marrow. Numbers of circulating RBCs are affected by changes in plasma volume, rate of RBC destruction or loss, splenic contraction, erythropoietin (EPO) secretion, and the rate of bone marrow production. A normal PCV is maintained by an endocrine loop that involves generation and release of erythropoietin (EPO) from the kidney in response to renal hypoxia. Erythropoietin stimulates platelet production as well as red cell production. However, erythropoietin does not stimulate white blood cell (WBC) production. Erythropoiesis and RBC numbers are also affected by hormones from the adrenal cortex, thyroid, ovary, testis, and anterior pituitary. Destruction Red cells have a finite circulating lifespan. In dogs, the average normal red cell circulates approximately 100 days. -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets. -
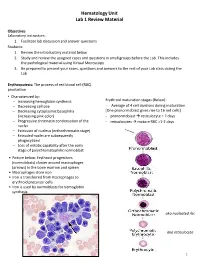
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Histology Histology
HISTOLOGY HISTOLOGY ОДЕСЬКИЙ НАЦІОНАЛЬНИЙ МЕДИЧНИЙ УНІВЕРСИТЕТ THE ODESSA NATIONAL MEDICAL UNIVERSITY Áiáëiîòåêà ñòóäåíòà-ìåäèêà Medical Student’s Library Серія заснована в 1999 р. на честь 100-річчя Одеського державного медичного університету (1900–2000 рр.) The series is initiated in 1999 to mark the Centenary of the Odessa State Medical University (1900–2000) 1 L. V. Arnautova O. A. Ulyantseva HISTÎLÎGY A course of lectures A manual Odessa The Odessa National Medical University 2011 UDC 616-018: 378.16 BBC 28.8я73 Series “Medical Student’s Library” Initiated in 1999 Authors: L. V. Arnautova, O. A. Ulyantseva Reviewers: Professor V. I. Shepitko, MD, the head of the Department of Histology, Cytology and Embryology of the Ukrainian Medical Stomatologic Academy Professor O. Yu. Shapovalova, MD, the head of the Department of Histology, Cytology and Embryology of the Crimean State Medical University named after S. I. Georgiyevsky It is published according to the decision of the Central Coordinational Methodical Committee of the Odessa National Medical University Proceedings N1 from 22.09.2010 Навчальний посібник містить лекції з гістології, цитології та ембріології у відповідності до програми. Викладено матеріали теоретичного курсу по всіх темах загальної та спеціальної гістології та ембріології. Посібник призначений для підготовки студентів до практичних занять та ліцензійного екзамену “Крок-1”. Arnautova L. V. Histology. A course of lectures : a manual / L. V. Arnautova, O. A. Ulyantseva. — Оdessa : The Оdessa National Medical University, 2010. — 336 p. — (Series “Medical Student’s Library”). ISBN 978-966-443-034-7 The manual contains the lecture course on histology, cytology and embryol- ogy in correspondence with the program. -

Complete Blood Count in Primary Care
Complete Blood Count in Primary Care bpac nz better medicine Editorial Team bpacnz Tony Fraser 10 George Street Professor Murray Tilyard PO Box 6032, Dunedin Clinical Advisory Group phone 03 477 5418 Dr Dave Colquhoun Michele Cray free fax 0800 bpac nz Dr Rosemary Ikram www.bpac.org.nz Dr Peter Jensen Dr Cam Kyle Dr Chris Leathart Dr Lynn McBain Associate Professor Jim Reid Dr David Reith Professor Murray Tilyard Programme Development Team Noni Allison Rachael Clarke Rebecca Didham Terry Ehau Peter Ellison Dr Malcolm Kendall-Smith Dr Anne Marie Tangney Dr Trevor Walker Dr Sharyn Willis Dave Woods Report Development Team Justine Broadley Todd Gillies Lana Johnson Web Gordon Smith Design Michael Crawford Management and Administration Kaye Baldwin Tony Fraser Kyla Letman Professor Murray Tilyard Distribution Zane Lindon Lyn Thomlinson Colleen Witchall All information is intended for use by competent health care professionals and should be utilised in conjunction with © May 2008 pertinent clinical data. Contents Key points/purpose 2 Introduction 2 Background ▪ Haematopoiesis - Cell development 3 ▪ Limitations of reference ranges for the CBC 4 ▪ Borderline abnormal results must be interpreted in clinical context 4 ▪ History and clinical examination 4 White Cells ▪ Neutrophils 5 ▪ Lymphocytes 9 ▪ Monocytes 11 ▪ Basophils 12 ▪ Eosinophils 12 ▪ Platelets 13 Haemoglobin and red cell indices ▪ Low haemoglobin 15 ▪ Microcytic anaemia 15 ▪ Normocytic anaemia 16 ▪ Macrocytic anaemia 17 ▪ High haemoglobin 17 ▪ Other red cell indices 18 Summary Table 19 Glossary 20 This resource is a consensus document, developed with haematology and general practice input. We would like to thank: Dr Liam Fernyhough, Haematologist, Canterbury Health Laboratories Dr Chris Leathart, GP, Christchurch Dr Edward Theakston, Haematologist, Diagnostic Medlab Ltd We would like to acknowledge their advice, expertise and valuable feedback on this document. -

Red Blood Cell Morphology in Patients with Β-Thalassemia Minor
J Lab Med 2017; 41(1): 49–52 Short Communication Carolin Körber, Albert Wölfler, Manfred Neubauer and Christoph Robier* Red blood cell morphology in patients with β-thalassemia minor DOI 10.1515/labmed-2016-0052 Keywords: β-thalassemia minor; erythrocytes; red blood Received July 11, 2016; accepted October 20, 2016; previously published cells; red blood cell morphology. online December 10, 2016 Abstract In β-thalassemias, the examination of a peripheral blood (PB) smear may provide relevant clues to initial diagnosis. Background: A systematic analysis of the occurrence of Complete laboratory investigation consists of the determina- red blood cell (RBC) abnormalities in β-thalassemia minor tion of the complete blood count, assessment of red blood has not been performed to date. This study aimed to iden- cell (RBC) morphology, high performance liquid chroma- tify and quantify the frequency of RBC abnormalities in tography (HPLC), hemoglobin electrophoresis and, where patients with β-thalassemia minor. necessary, DNA analysis [1]. Especially in the clinically Methods: We examined blood smears of 33 patients with severe forms referred to as β-thalassemia major and interme- β-thalassemia minor by light microscopy for the occur- dia, RBC abnormalities are often markedly apparent [2]. In rence of 15 defined RBC abnormalities. In the case of posi- β-thalassemia minor, also called β-thalassemia trait, the car- tivity, the abnormal cells/20 high power fields (HPF) at riers are usually clinically asymptomatic, showing persistent 1000-fold magnification were counted. microcytosis and hypochromia or mild microcytic anemia [1, Results: Anisocytosis, poikilocytosis and target cells 3]. The PB smear may show microcytosis, hypochromia and, (median 42/20 HPF) were observed in all, and ovalocytes infrequently, poikilocytosis [2]. -

Laboratory Diagnosis Review
Laboratory Diagnosis Review Hematology Definition: The study of the three cellular elements of blood: Red Blood Cells (RBCs), White Blood Cells (WBCs), and Platelets Hemoglobin (Hgb or Hb): The oxygen carrying compound in RBCs Reference Range: Men 14-18 g/dL, Women 12-16 g/dL, Boy and girl levels are equal till age 11 Smoking increases, Pregnancy decreases, Capillary levels in newborns are higher than venous levels, Race, Position, and Time of day have minor effects, High WBCs may falsely raise Hgb. Below normal Hgb = anemia Red Blood Cell Count Reference Range: Men 4l5-6 million / cubic ml, Women4.0-5.5 million / ml3. Hematocrit (Hct): The ratio of RBCs to plasma Reference Range, Men 40%-54%, Women 37%-47%, Depends mostly on the number of RBCs but is slightly effected by the average RBC size, Not measured directly, but is calculated from the RBC count and the mean corpuscular volume (MCV). Increased by smoking, Decrease = anemia Useful Relationships: Hb X 3 = Hct RBCs (millions) X 3 = Hgb RBCs (millions) X 9 = Hct Wintrobe Indices: These indices are only significant if the RBCs, Hgb, and/or Hct. is abnormal MCV: Mean Corpuscular Volume MCH: Mean Corpuscular Hemoglobin MCHC: Mean Corpuscular Hemoglobin Concentration RDW: Red blood cell Distribution Width MCV: Reference Ranges: Men 80-95 fl (femtoliters), Women 81-99 fl (femto = 1 quadrillionth) Increased MCV = Macrocytosis, Decreased = Microcytosis MCV is increased by smoking, by B12 and/or folic acid deficiency, chronic liver disease, chronic alcoholism, Cardiorespiratory problems… Some macrocytic patients will not have macrocytosis MCV is decreased by iron deficiency, thalassemia, and anemia of chronic disease MCH: Reference Range 27-31 pg MCH is increased by Macrocytic anemias. -

10 11 Cyto Slides 81-85
NEW YORK STATE CYTOHEMATOLOGY PROFICIENCY TESTING PROGRAM Glass Slide Critique ~ November 2010 Slide 081 Diagnosis: MDS to AML 9 WBC 51.0 x 10 /L 12 Available data: RBC 3.39 x 10 /L 72 year-old female Hemoglobin 9.6 g/dL Hematocrit 29.1 % MCV 86.0 fL Platelet count 16 x 109 /L The significant finding in this case of Acute Myelogenous Leukemia (AML) was the presence of many blast forms. The participant median for blasts, all types was 88. The blast cells in this case (Image 081) are large, irregular in shape and contain large prominent nucleoli. It is difficult to identify a blast cell as a myeloblast without the presence of an Auer rod in the cytoplasm. Auer rods were reported by three participants. Two systems are used to classify AML into subtypes, the French- American-British (FAB) and the World Health Organization (WHO). Most are familiar with the FAB classification. The WHO classification system takes into consideration prognostic factors in classifying AML. These factors include cytogenetic test results, patient’s age, white blood cell count, pre-existing blood disorders and a history of treatment with chemotherapy and/or radiation therapy for a prior cancer. The platelet count in this case was 16,000. Reduced number of platelets was correctly reported by 346 (94%) of participants. Approximately eight percent of participants commented that the red blood cells in this case were difficult to evaluate due to the presence of a bluish hue around the red blood cells. Comments received included, “On slide 081 the morphology was difficult to evaluate since there was a large amount of protein surrounding RBC’s”, “Slide 081 unable to distinguish red cell morphology due to protein” and “Unable to adequately assess morphology on slide 081 due to poor stain”. -

Advanced Blood Cell Id: Peripheral Blood Findings in Sickle Cell Anemia
ADVANCED BLOOD CELL ID: PERIPHERAL BLOOD FINDINGS IN SICKLE CELL ANEMIA Educational commentary is provided for participants enrolled in program #259- Advanced Blood Cell Identification. This virtual blood cell identification program includes case studies with more difficult challenges. To view the blood cell images in more detail, click on the sample identification numbers underlined in the paragraphs below. This will open a virtual image of the selected cell and the surrounding fields. If the image opens in the same window as the commentary, saving the commentary PDF and opening it outside your browser will allow you to switch between the commentary and the images more easily. Click on this link for the API ImageViewerTM Instructions. Learning Outcomes After completing this exercise, participants should be able to: • describe morphologic features of normal peripheral blood leukocytes. • identify morphologic characteristics distinctive of sickle cells. • distinguish selected RBC inclusions based on morphologic features. • describe significant morphologic characteristics of nucleated red blood cells. Case Study The CBC from a 30 year old African American male is as follows: WBC=9.5 x 109/L, RBC=1.66 x 1012/L, Hgb=5.0 g/dL, Hct=13.9%, MCV=83.7 fL, MCH=30.1 pg, MCHC=36.0 g/dL, RDW-CV=24.9%, MPV=9.6 fL, Platelet=326 x 109/L. Educational Commentary The cells and RBC inclusions chosen for identification in this testing event were seen in the peripheral blood of a man with a severe anemia resulting from sickle cell disease. The cell shown in ABI-08 contains a Howell-Jolly body. -

© 2016 First Aid for the USMLE Step 1
Index A Abscess, 442 vs. aspirin, in pediatric patients, 446 Achondroplasia, 426 Abacavir, 184 Absence seizures, 494 free radical injury and, 221 autosomal dominance of, 71 for HIV, 186 drug therapy for, 500 necrosis caused by, 252 chromosome associated with, 75 Abciximab, 214, 407 treatments for, 638 for osteoarthritis, 430 endochondral ossification in, 425 thrombogenesis and, 385 Absolute risk reduction (ARR), 34, for tension headaches, 494 AChR (acetylcholine receptor), 229 Abdominal aorta, 342 646 toxicity effects, 446 Acid-base physiology, 543 atherosclerosis in, 286, 645 Absorption disorders, anemia caused toxicity treatment for, 251 Acidemia, 543 bifurcation of, 609 by, 388 Acetazolamide, 254, 557 diuretic effect on, 558 498 92 Abdominal aortic aneurysm, 286 Abuse for glaucoma, Acidic amino acids, metabolic acidosis caused by, 543 Acidosis, 543 Abdominal colic confidentiality exceptions and, 41 in nephron physiology, 537 contractility in, 267 lead poisoning as cause, 389 dependent personality disorder for pseudotumor cerebri, 471 hyperkalemia caused by, 542 Abdominal distension and, 519 site of action, 556 Acid phosphatase, in neutrophils, 378 duodenal atresia as cause, 338 Acalculia, 464 Acetoacetate, metabolism of, 102 Acid reflux Abdominal pain Acamprosate Acetone breath, in diabetic esophageal strictures and, 354 Budd-Chiari syndrome as for alcoholism, 523, 638 ketoacidosis, 331 esophagitis and, 354 cause, 368, 630 diarrhea caused by, 252 Acetylation, 57 H blockers for, 374 cilostazol/dipyridamole as Acanthocytes, 386 2 Acetylcholine -

Research Article
z Available online at http://www.journalcra.com INTERNATIONAL JOURNAL OF CURRENT RESEARCH International Journal of Current Research Vol. 9, Issue, 12, pp.62497-62502, December, 2017 ISSN: 0975-833X RESEARCH ARTICLE INCLUSION BODIES - A REVIEW 1Dr. Mrunalini Mahesh Dadpe, 2, *Dr. Sourab Kumar, 3Dr. Mahesh Dadpe, 4Dr. Payoshnee Bhalinge Jadhav, 5Dr. Abhishek Jadhav and 6Dr. Shilpi Suman 1, 4Department of Oral Pathology, M A Rangoonwala Dental College, Aazam Campus, Camp, Pune 411001, India 2, 5, 6Department of oral Pathology, D.Y. Patil School of Dentistry, Nerul, Navi Mumbai 400706, India 3MIDSR Dental College, Vishwanathpuram, Amba Jogai Road, Latur 413531, India ARTICLE INFO ABSTRACT Article History: The word inclusion means incorporation. Inclusion bodies are nuclear or cytoplasmic aggregates of Received 16th September, 2017 stainable substances which are usually ‘proteins’. They typically represent sites of viral multiplication Received in revised form in bacteria or in a eukaryotic cell and usually consist of viral capsid proteins. Inclusion bodies are 17th October, 2017 typically identified within a cell by their different appearance. Accepted 25th November, 2017 Published online 27th December, 2017 Key words: Proteins, Viral, Types, Structures Copyright © 2017, Mrunalini Mahesh Dadpe et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Citation: Dr. Mrunalini Mahesh Dadpe, Dr. Sourab Kumar, Dr. Mahesh Dadpe, Dr. Payoshnee Bhalinge Jadhav, Dr. Abhishek Jadhav and Dr. Shilpi Suman, 2017. “Inclusion Bodies - A Review”, International Journal of Current Research, 9, (12), 62497-62502. Classification INTRODUCTION Viral inclusion bodies Inclusion bodies (IB) can also be hallmarks of genetic diseases, as in the case of Neuronal Inclusion bodies in neural disorders, A.