E-Learn LAB — RCD 1708
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hemolytic Disease of the Newborn
Intensive Care Nursery House Staff Manual Hemolytic Disease of the Newborn INTRODUCTION and DEFINITION: Hemolytic Disease of the Newborn (HDN), also known as erythroblastosis fetalis, isoimmunization, or blood group incompatibility, occurs when fetal red blood cells (RBCs), which possess an antigen that the mother lacks, cross the placenta into the maternal circulation, where they stimulate antibody production. The antibodies return to the fetal circulation and result in RBC destruction. DIFFERENTIAL DIAGNOSIS of hemolytic anemia in a newborn infant: -Isoimmunization -RBC enzyme disorders (e.g., G6PD, pyruvate kinase deficiency) -Hemoglobin synthesis disorders (e.g., alpha-thalassemias) -RBC membrane abnormalities (e.g., hereditary spherocytosis, elliptocytosis) -Hemangiomas (Kasabach Merritt syndrome) -Acquired conditions, such as sepsis, infections with TORCH or Parvovirus B19 (anemia due to RBC aplasia) and hemolysis secondary to drugs. ISOIMMUNIZATION A. Rh disease (Rh = Rhesus factor) (1) Genetics: Rh positive (+) denotes presence of D antigen. The number of antigenic sites on RBCs varies with genotype. Prevalence of genotype varies with the population. Rh negative (d/d) individuals comprise 15% of Caucasians, 5.5% of African Americans, and <1% of Asians. A sensitized Rh negative mother produces anti-Rh IgG antibodies that cross the placenta. Risk factors for antibody production include 2nd (or later) pregnancies*, maternal toxemia, paternal zygosity (D/D rather than D/d), feto-maternal compatibility in ABO system and antigen load. (2) Clinical presentation of HDN varies from mild jaundice and anemia to hydrops fetalis (with ascites, pleural and pericardial effusions). Because the placenta clears bilirubin, the chief risk to the fetus is anemia. Extramedullary hematopoiesis (due to anemia) results in hepatosplenomegaly. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

Clinical Pathology Interpretation Barbara Horney
CLINICAL PATHOLOGY PATHOLOGIE CLINIQUE Clinical pathology interpretation Barbara Horney History, physical examination, and Table 1. Hematologic findings from a lethargic, laboratory findings 4-year-old schipperke 4-year-old, spayed female, schipperke was pre- Blood cell count Reference range A sented because of mild lethargy. Pale mucous mem- White blood cells branes were observed on physical examination. Table 1 (WBC) gives the results of the hematological examination of Total 6.0 X 109/L 6.0-17.1 X 109/L blood at Differential samples taken this time. No significant abnor- segmented 65% 3.85 X 109/L 3.6-11.5 X 109/L malities were identified on the serum biochemical neutrophils profile. eosinophils 2% 0.12 X 109/L 0.01-1.25 X 109/L lymphocytes 27% 1.59 X 109/L 1.0-4.8 X 109/L Interpretation and discussion monocytes 6% 0.35 X 109/L 0.15-1.35 X 109/L Red blood cells The hematology results can be summarized as severe, Total 1.2 X 1012/L 5.5-8.5 X 109/L microcytic, normochromic, nonregenerative anemia nucleated 1/100 WBC <1-2 per 100 WBC associated with marked spherocytosis. spherocytes 4+ microcytosis 2+ The presence of spherocytes is often associated with immune-mediated hemolytic disease [1,2], although Platelets estimated normal hereditary membrane defects [3] and zinc toxicosis [4] in number can also result in spherocyte formation. A direct antibody Reticulocytes 0 X 109/L up to 120 X 109/L test (Coomb's test) was weakly positive. This finding can Hemoglobin 22 g/L 120-180 g/L support the tentative diagnosis of anemia of immune- Hematocrit 0.068 L/L 0.37-0.55 L/L mediated etiology, although this test is subject to both Mean corpuscular false positive and false negative results [2,5]. -

Molecular Basis of Spectrin Deficiency in Beta Spectrin Durham. a Deletion
Molecular basis of spectrin deficiency in beta spectrin Durham. A deletion within beta spectrin adjacent to the ankyrin-binding site precludes spectrin attachment to the membrane in hereditary spherocytosis. H Hassoun, … , S S Chiou, J Palek J Clin Invest. 1995;96(6):2623-2629. https://doi.org/10.1172/JCI118327. Research Article We describe a spectrin variant characterized by a truncated beta chain and associated with hereditary spherocytosis. The clinical phenotype consists of a moderate hemolytic anemia with striking spherocytosis and mild spiculation of the red cells. We describe the biochemical characteristics of this truncated protein which constitutes only 10% of the total beta spectrin present on the membrane, resulting in spectrin deficiency. Analysis of reticulocyte cDNA revealed the deletion of exons 22 and 23. We show, using Southern blot analysis, that this truncation results from a 4.6-kb genomic deletion. To elucidate the basis for the decreased amount of the truncated protein on the membrane and the overall spectrin deficiency, we show that (a) the mutated gene is efficiently transcribed and its mRNA abundant in reticulocytes, (b) the mutant protein is normally synthesized in erythroid progenitor cells, (c) the stability of the mutant protein in the cytoplasm of erythroblasts parallels that of the normal beta spectrin, and (d) the abnormal protein is inefficiently incorporated into the membrane of erythroblasts. We conclude that the truncation within the beta spectrin leads to inefficient incorporation of the mutant protein into the skeleton despite its normal synthesis and stability. We postulate that this misincorporation results from conformational changes of the beta spectrin subunit affecting the binding of the abnormal heterodimer to ankyrin, and we provide evidence […] Find the latest version: https://jci.me/118327/pdf Molecular Basis of Spectrin Deficiency in p8 Spectrin Durham A Deletion within .3 Spectrin Adjacent to the Ankyrin-binding Site Precludes Spectrin Attachment to the Membrane in Hereditary Spherocytosis Hani Hassoun,* John N. -

Erythrocytes: Overview, Morphology, Quantity by AH Rebar Et
In: A Guide to Hematology in Dogs and Cats, Rebar A.H., MacWilliams P.S., Feldman B.F., Metzger F.L., Pollock R.V.H. and Roche J. (Eds.). Publisher: Teton NewMedia, Jackson WY (www.veterinarywire.com). Internet Publisher: International Veterinary Information Service, Ithaca NY (www.ivis.org), 8-Feb-2005; A3304.0205 Erythrocytes: Overview, Morphology, Quantity A.H. Rebar1, P.S. MacWilliams2, B.F. Feldman 3, F.L. Metzger 4, R.V.H. Pollock 5 and J. Roche 6 1Dept of Veterinary Pathobiology, School of Veterinary Medicine, Purdue University, IN,USA. 2Dept of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, WI, USA. 3Dept of Biomedical Sciences & Pathobiology, VA-MD - Regional College of Veterinary Medicine, Virginia Tech, VA, USA. 4Metzger Animal Hospital,State College,PA, USA. 5Fort Hill Company, Montchanin, DE, USA. 6 Hematology Systems, IDEXX Laboratories, Westbrook, ME, USA. Overview Production Red blood cells (RBC) are produced in the bone marrow. Numbers of circulating RBCs are affected by changes in plasma volume, rate of RBC destruction or loss, splenic contraction, erythropoietin (EPO) secretion, and the rate of bone marrow production. A normal PCV is maintained by an endocrine loop that involves generation and release of erythropoietin (EPO) from the kidney in response to renal hypoxia. Erythropoietin stimulates platelet production as well as red cell production. However, erythropoietin does not stimulate white blood cell (WBC) production. Erythropoiesis and RBC numbers are also affected by hormones from the adrenal cortex, thyroid, ovary, testis, and anterior pituitary. Destruction Red cells have a finite circulating lifespan. In dogs, the average normal red cell circulates approximately 100 days. -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

20 Hemolytic Anemias Due to Abnormal Red Cell Enzymes
Hemolytic Anemias Due to Abnormal Red Cell Enzymes MODULE Hematology and Blood Bank Technique 20 HEMOLYTIC ANEMIAS DUE TO Notes ABNORMAL RED CELL ENZYMES 20.1 INTRODUCTION The main metabolic substrate for the RBCs is glucose. It is metabolized by two pathways: approximately 90% of the glucose is metabolized through the Embden Meyerhoff (glycolytic) pathway and the rest by the hexose monophosphate (HMP) pathway. In the Embden Meyerhoff (glycolytic) pathway glucose is metabolized to lactate through a series of enzymatic steps. Each molecule of glucose gives rise to 2 molecules of ATP. The ATP provides energy to maintain red cell volume, shape and flexibility. An ATP dependent pump in the red cell membrane actively keeps sodium out of the cell and potassium inside. The red cell has the enzymes that are needed for the glycolytic pathway. These enzymes help break down glucose to generate ATP which is the source of energy. About 10% of the glucose is diverted to the Hexose Monophosphate shunt pathway and this is essential for protection of red cells from oxidative stress. This pathway is necessary for the generation of NADPH which then reduces oxidized glutathione (GSSG) to reduced glutathione (GSH). GSH prevents the accumulation of H2O2 and the oxidation of hemoglobin to methemoglobin. When the level of GSH falls, H2O2 accumulates in the cell and oxidizes the hemoglobin to methemoglobin which becomes denatured and precipitates as Heinz bodies. These inclusions are rigid and attached to the red cell membrane and make the red cell susceptible to hemolysis. The NADPH required in this pathway is generated by the enzyme Glucose 6 phosphate dehydrogenase (G6PD). -
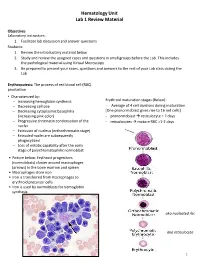
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Rbcs) in Both the Sinus and Cord, Histiocytes Werc Swollen by a Granu}Ar Substance in the Cytoplasm and Also Many Secondary Lysosomes
The UOEHAssociationUOEH Association ofofHealth Health Sciences JUOEH20(1)!11-19 (1998) 11 [Original) Pathological Study Splenomegaly Associated with Cadmium-inducedof Anemia in Rats Tetsuo HAMADA', Akihide TANIMOT02, Nobuyuki ARIMA!, Yoshihiro IDEL, Takakazu SASAGURI2, Shohei SHIMiVIRI2, Yoshitaka MURATA', Ke-Yong WANG2 and Yasuyuki SASAGURIL' 'Dopartment of'Surgical Pathotogy, University llosPital ?Dopartment qf Palhology and Cell Biotogy, Scheol of' Adlrdicine, (1itib'ersity of Occapational and Environmental Health., .lapan. }'dhatanishi-ku, Kiialp,ztshu 807-8imr, .1apan Ahstract : Splenomegaly was observcd both in male and R)malc Spraguc-Dawley rats after 1 week of exposure to CdC12 (O,6 mg (:d/kg/day), Sp]een weight reached about double that in controls by 8 weeks of Cd exposure. Histopathologica] cxamination of the enlargcd spleen rcvcaled that iron- and lipid-laden histiocytes were clustered in tha periarterial lymphatic sheath, and the red pulp appeared to be expanded. It is noteworthy that electron microscopy rcvealed markcd poikilocytosis and Hcinz body formation in red blood cells (RBCs) in both the sinus and cord, Histiocytes werc swollen by a granu}ar substance in the cytoplasm and also many secondary lysosomes. Thesc morphological findings indicatc that degradation of damagcd RBCs induced by exposure to Cd might bc promoted in the splccn and possibly cause splenomcgaly, This RBC damage-hcmolysis-splcnomegaly sequence is also considered to be associated with the etioiogy of Cd-induced anemia, In addition to the abnormal RBC degradation, nuclel of ]ymphocytes in thc Cd-cxposcd spleen exhibited high elcctron density, consistent with a preapoptotic statc suggesting the immunosupprcssive effhct ofCd. Kay woralf :spienomegaly, poiki]ocytosis, Heinz body, cadmiurn, anemia, (Received 18 November 1997, accepted I9January 1998) Introduction Cd was reported to cause anemia in dogs as early as 1896 [1], and subsequently, anemia was found in humans after Cd exposure [2]. -

Hereditary Spherocytosis
Hereditary Spherocytosis o RBC band 3 protein testing is a very sensitive and Indications for Ordering specific test for the diagnosis of hereditary Use to confirm diagnosis of hereditary spherocytosis when spherocytosis hemolytic anemia and spherocytes are present Physiology • RBC band 3 protein is a major structural protein of RBCs Test Description o Reduction in the amount of band 3 fluorescence after Test Methodology binding with EMA correlates with spherocytosis • Red blood cell (RBC) surface protein band 3 staining with Genetics eosin-5-maleimide (EMA) analyzed by flow cytometry Clinical Validation Genes: ANK1, EPB42, SLC4A1, SPTA1, SPTB • Validated against the clinical diagnosis of hereditary Inheritance spherocytosis supported by osmotic fragility and/or • Autosomal dominant: 75% molecular testing • Autosomal recessive: 25% Tests to Consider Penetrance: variable Structure/Function Primary Test • Chromosomal location: 17q21.31 RBC Band 3 Protein Reduction in Hereditary Spherocytosis • Provides structure for the red cell cytoskeleton 2008460 • Use to confirm diagnosis of hereditary spherocytosis Test Interpretation when hemolytic anemia and spherocytes are present Sensitivity/Specificity Related Test • Clinical sensitivity: 93% Osmotic Fragility, Erythrocyte 2002257 • Analytical sensitivity/specificity: unknown • Functional testing of RBC sensitivity to osmotic stress Results Disease Overview • Normal o Normal staining of band 3 protein with EMA does not Prevalence: 1/2,000 in northern Europeans suggest hereditary spherocytosis -

Complete Blood Count in Primary Care
Complete Blood Count in Primary Care bpac nz better medicine Editorial Team bpacnz Tony Fraser 10 George Street Professor Murray Tilyard PO Box 6032, Dunedin Clinical Advisory Group phone 03 477 5418 Dr Dave Colquhoun Michele Cray free fax 0800 bpac nz Dr Rosemary Ikram www.bpac.org.nz Dr Peter Jensen Dr Cam Kyle Dr Chris Leathart Dr Lynn McBain Associate Professor Jim Reid Dr David Reith Professor Murray Tilyard Programme Development Team Noni Allison Rachael Clarke Rebecca Didham Terry Ehau Peter Ellison Dr Malcolm Kendall-Smith Dr Anne Marie Tangney Dr Trevor Walker Dr Sharyn Willis Dave Woods Report Development Team Justine Broadley Todd Gillies Lana Johnson Web Gordon Smith Design Michael Crawford Management and Administration Kaye Baldwin Tony Fraser Kyla Letman Professor Murray Tilyard Distribution Zane Lindon Lyn Thomlinson Colleen Witchall All information is intended for use by competent health care professionals and should be utilised in conjunction with © May 2008 pertinent clinical data. Contents Key points/purpose 2 Introduction 2 Background ▪ Haematopoiesis - Cell development 3 ▪ Limitations of reference ranges for the CBC 4 ▪ Borderline abnormal results must be interpreted in clinical context 4 ▪ History and clinical examination 4 White Cells ▪ Neutrophils 5 ▪ Lymphocytes 9 ▪ Monocytes 11 ▪ Basophils 12 ▪ Eosinophils 12 ▪ Platelets 13 Haemoglobin and red cell indices ▪ Low haemoglobin 15 ▪ Microcytic anaemia 15 ▪ Normocytic anaemia 16 ▪ Macrocytic anaemia 17 ▪ High haemoglobin 17 ▪ Other red cell indices 18 Summary Table 19 Glossary 20 This resource is a consensus document, developed with haematology and general practice input. We would like to thank: Dr Liam Fernyhough, Haematologist, Canterbury Health Laboratories Dr Chris Leathart, GP, Christchurch Dr Edward Theakston, Haematologist, Diagnostic Medlab Ltd We would like to acknowledge their advice, expertise and valuable feedback on this document. -

Red Blood Cell Morphology in Patients with Β-Thalassemia Minor
J Lab Med 2017; 41(1): 49–52 Short Communication Carolin Körber, Albert Wölfler, Manfred Neubauer and Christoph Robier* Red blood cell morphology in patients with β-thalassemia minor DOI 10.1515/labmed-2016-0052 Keywords: β-thalassemia minor; erythrocytes; red blood Received July 11, 2016; accepted October 20, 2016; previously published cells; red blood cell morphology. online December 10, 2016 Abstract In β-thalassemias, the examination of a peripheral blood (PB) smear may provide relevant clues to initial diagnosis. Background: A systematic analysis of the occurrence of Complete laboratory investigation consists of the determina- red blood cell (RBC) abnormalities in β-thalassemia minor tion of the complete blood count, assessment of red blood has not been performed to date. This study aimed to iden- cell (RBC) morphology, high performance liquid chroma- tify and quantify the frequency of RBC abnormalities in tography (HPLC), hemoglobin electrophoresis and, where patients with β-thalassemia minor. necessary, DNA analysis [1]. Especially in the clinically Methods: We examined blood smears of 33 patients with severe forms referred to as β-thalassemia major and interme- β-thalassemia minor by light microscopy for the occur- dia, RBC abnormalities are often markedly apparent [2]. In rence of 15 defined RBC abnormalities. In the case of posi- β-thalassemia minor, also called β-thalassemia trait, the car- tivity, the abnormal cells/20 high power fields (HPF) at riers are usually clinically asymptomatic, showing persistent 1000-fold magnification were counted. microcytosis and hypochromia or mild microcytic anemia [1, Results: Anisocytosis, poikilocytosis and target cells 3]. The PB smear may show microcytosis, hypochromia and, (median 42/20 HPF) were observed in all, and ovalocytes infrequently, poikilocytosis [2].