How I Manage Patients with Atypical Microcytic Anaemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Clinical Pathology Interpretation Barbara Horney
CLINICAL PATHOLOGY PATHOLOGIE CLINIQUE Clinical pathology interpretation Barbara Horney History, physical examination, and Table 1. Hematologic findings from a lethargic, laboratory findings 4-year-old schipperke 4-year-old, spayed female, schipperke was pre- Blood cell count Reference range A sented because of mild lethargy. Pale mucous mem- White blood cells branes were observed on physical examination. Table 1 (WBC) gives the results of the hematological examination of Total 6.0 X 109/L 6.0-17.1 X 109/L blood at Differential samples taken this time. No significant abnor- segmented 65% 3.85 X 109/L 3.6-11.5 X 109/L malities were identified on the serum biochemical neutrophils profile. eosinophils 2% 0.12 X 109/L 0.01-1.25 X 109/L lymphocytes 27% 1.59 X 109/L 1.0-4.8 X 109/L Interpretation and discussion monocytes 6% 0.35 X 109/L 0.15-1.35 X 109/L Red blood cells The hematology results can be summarized as severe, Total 1.2 X 1012/L 5.5-8.5 X 109/L microcytic, normochromic, nonregenerative anemia nucleated 1/100 WBC <1-2 per 100 WBC associated with marked spherocytosis. spherocytes 4+ microcytosis 2+ The presence of spherocytes is often associated with immune-mediated hemolytic disease [1,2], although Platelets estimated normal hereditary membrane defects [3] and zinc toxicosis [4] in number can also result in spherocyte formation. A direct antibody Reticulocytes 0 X 109/L up to 120 X 109/L test (Coomb's test) was weakly positive. This finding can Hemoglobin 22 g/L 120-180 g/L support the tentative diagnosis of anemia of immune- Hematocrit 0.068 L/L 0.37-0.55 L/L mediated etiology, although this test is subject to both Mean corpuscular false positive and false negative results [2,5]. -

Impaired Lysosomal Acidification Triggers Iron Deficiency And
RESEARCH ARTICLE Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo King Faisal Yambire1, Christine Rostosky2, Takashi Watanabe3, David Pacheu-Grau1, Sylvia Torres-Odio4, Angela Sanchez-Guerrero1,2, Ola Senderovich5, Esther G Meyron-Holtz5, Ira Milosevic2, Jens Frahm3, A Phillip West4, Nuno Raimundo1* 1Institute of Cellular Biochemistry, University Medical Center Goettingen, Goettingen, Germany; 2European Neuroscience Institute, a Joint Initiative of the Max-Planck Institute and of the University Medical Center Goettingen, Goettingen, Germany; 3Biomedizinische NMR, Max-Planck Institute for Biophysical Chemistry, Goettingen, Germany; 4Department of Microbial Pathogenesis and Immunology, Texas A&M University Health Science Center, Austin, United States; 5Faculty of Biotechnology and Food Engineering, Technion Israel Institute of Technology, Haifa, Israel Abstract Lysosomal acidification is a key feature of healthy cells. Inability to maintain lysosomal acidic pH is associated with aging and neurodegenerative diseases. However, the mechanisms elicited by impaired lysosomal acidification remain poorly understood. We show here that inhibition of lysosomal acidification triggers cellular iron deficiency, which results in impaired mitochondrial function and non-apoptotic cell death. These effects are recovered by supplying iron via a lysosome-independent pathway. Notably, iron deficiency is sufficient to trigger inflammatory signaling in cultured primary neurons. Using a mouse model of impaired lysosomal acidification, we observed a robust iron deficiency response in the brain, verified by in vivo magnetic resonance *For correspondence: imaging. Furthermore, the brains of these mice present a pervasive inflammatory signature [email protected] associated with instability of mitochondrial DNA (mtDNA), both corrected by supplementation of goettingen.de the mice diet with iron. Our results highlight a novel mechanism linking impaired lysosomal Competing interests: The acidification, mitochondrial malfunction and inflammation in vivo. -

Kawasaki Disease with Glucose-6-Phosphate Dehydrogenase Deficiency, Case Report
Saudi Pharmaceutical Journal (2014) xxx, xxx–xxx King Saud University Saudi Pharmaceutical Journal www.ksu.edu.sa www.sciencedirect.com CASE REPORT Kawasaki disease with Glucose-6-Phosphate Dehydrogenase deficiency, case report Hesham Radi Obeidat a,*, Sahar Al-Dossary b, Abdulsalam Asseri a a Pharmacy Department, Saad Specialist Hospital, Alkhobar 31952, Saudi Arabia b Pediatric and Neonatology Department, Saad Specialist Hospital, Alkhobar 31952, Saudi Arabia Received 28 August 2014; accepted 11 November 2014 KEYWORDS Abstract Kawasaki disease (KD) is an acute, self-limited vasculitis of unknown etiology that Kawasaki disease; occurs predominantly in infants and children younger than 5 years of age. Coronary artery abnor- G6PD; malities are the most serious complication. Aspirin Based on the literatures infusion of Intravenous Immunoglobulin of 2 g/kg and a high dose of oral aspirin up to 100 mg/kg/day are the standard treatment for Kawasaki disease in the acute stage, and should be followed by antiplatelet dose of aspirin for thrombocytosis. Glucose-6-Phos- phate Dehydrogenase (G6PD) deficiency is an inherited X-linked hereditary disorder, and aspirin can induce hemolysis in patients with G6PD deficiency. We report a case of a 5 year and 8 month old male with KD and G6PD deficiency. ª 2014 The Authors. Production and hosting by Elsevier B.V. on behalf of King Saud University. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/). 1. Introduction genetic predisposition or infectious agents are likely to be the cause. Kawasaki disease was first described in 1967 by Tomisaku Recently, guidelines were published by the American Heart Kawasaki and has replaced acute rheumatic fever as the lead- Association (AHA) to aid in the diagnosis and management of ing cause of acquired heart disease among children in devel- Kawasaki disease. -

Newborn Screening Result for Bart's Hemoglobin
NEWBORN SCREENING RESULT FOR BART’S HEMOGLOBIN Physician’s information sheet developed by the Nebraska Newborn Screening Program with review by James Harper, MD, Pediatric Hematologist with UNMC Follow-up program, and member of the Nebraska Newborn Screening Advisory Committee. BACKGROUND The alpha thalassemias result from the loss of alpha globin genes. There are normally four genes for alpha globin production so that the loss of one to four genes is possible. The lack of one gene causes alpha thalassemia 2 (silent carrier) with no clinically detectable problems but may cause small amounts of hemoglobin Barts to be present in newborn blood samples. Alpha thalassemia trait (Alpha thalassemia 1) results from loss of two genes and causes a mild microcytic anemia which may resemble iron deficiency anemia. The loss of three genes causes hemoglobin H diseases which is a moderately severe form of thalassemia. The lack of all four genes causes hydrops fetalis and is usually fatal in utero. In general, only the loss of one or two genes is seen in African Americans. Individuals from Southeast Asia and the Mediterranean may have all four types of alpha thalassemia. The percentage of hemoglobin Barts in the blood sample may indicate the number of alpha genes that have been lost. However, the percentage of hemoglobin Barts is not directly measurable with the current methodology used by the newborn screening laboratory. Only the presence of Barts hemoglobin in relation to fetal and adult hemoglobin, and variants S, C, D and E can be detected. RECOMMENDED WORK UP In addition to the standard newborn hemoglobinopathy confirmation (hemoglobin electrophoresis), to separate those patients with alpha thalassemia silent carrier from the patients with alpha thalassemia trait, we recommend that these babies have the following labs drawn at their 6 month well baby check: CBC with retic count, ferritin, and a hemoglobin electropheresis. -

Hemolytic Anemia Caused by Hereditary Pyruvate Kinase Deficiency in a West Highland White Terrier Dog
Arch Med Vet 44, 195-200 (2012) COMMUNICATION Hemolytic anemia caused by hereditary pyruvate kinase deficiency in a West Highland White Terrier dog Anemia hemolítica causada por la deficiencia de piruvato quinasa hereditaria en un perro West Highland White Terrier NRC Hlavaca, LA Lacerdaa*, FO Conradob, PS Hünninga, M Seibertc, FHD Gonzálezd, U Gigere aPostgraduate Program in Veterinary Sciences, Universidade Federal Rio Grande do Sul, Porto Alegre, RS, Brasil. bVeterinary Clinic Pathology, Universidade Federal Rio Grande do Sul, Porto Alegre, RS, Brasil. cClinic Pathology, PetLab Ltda, Porto Alegre, Brasil. dDepartment of Veterinary Clinic Pathology, Faculty of Veterinary, Universidade Federal Rio Grande do Sul, Porto Alegre, RS, Brasil. eLaboratory of Genetic Diseases, University of Pennsilvania, Philadelphia, United States. RESUMEN La deficiencia de piruvato quinasa (PK) es un desorden hemolítico autosómico recesivo descrito en perros y gatos. La piruvato quinasa es una de las enzimas regulatorias esenciales de la glicólisis anaeróbica, la deficiencia de esta enzima causa una destrucción prematura de los eritrocitos. El presente es un estudio de caso y relata los hallazgos clínicos y paraclínicos en un perro brasileño de la raza West Highland White Terrier (WHWT) con historia de debilidad e intolerancia al ejercicio. El paciente presentaba mucosas pálidas, anemia hemolítica bastante regenerativa y osteoclerosis. La deficiencia de PK fue confirmada a través de una prueba de ADN raza específica para la inserción 6bp en el extremo 3’ del exón 10 de la secuencia del gen de la piruvato quinasa eritrocitaria (R-PK) como fue descrito. Al perro se le practicó eutanasia a los 20 meses de edad debido al deterioro de su estado clínico, el cual incluyó anemia e incompatibilidad sanguínea. -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets. -

Effect of Genotype on Micronutrient Absorption and Metabolism: a Review of Iron, Copper, Iodine and Selenium, and Folates Richard Mithen
Int. J. Vitam. Nutr. Res., 77 (3), 2007, 205–216 Effect of Genotype on Micronutrient Absorption and Metabolism: a Review of Iron, Copper, Iodine and Selenium, and Folates Richard Mithen Institute of Food Research, Colney Lane, Norwich, NR4 7UA, UK Received for publication: July 28, 2006 Abstract: For the majority of micronutrients, there are very little data, or none at all, on the role of genetic poly- morphisms on their absorption and metabolism. In many cases, the elucidation of biochemical pathways and regulators of homeostatic mechanisms have come from studies of individuals that have mutations in certain genes. Other polymorphisms in these genes that result in a less severe phenotype may be important in determining the natural range of variation in absorption and metabolism that is commonly observed. To illustrate some of these aspects, I briefly review the increased understanding of iron metabolism that has arisen from our knowledge of the effects of mutations in several genes, the role of genetic variation in mediating the nutritional effects of io- dine and selenium, and finally, the interaction between a genetic polymorphism in folate metabolism and folic acid fortification. Key words: Micronutrients, genetic polymorphisms, iron, iodine, selenium, folates Introduction the interpretation of epidemiological studies, in which some of the variation observed in nutrient status or re- Recently there has been considerable interest in the role quirement may be due to genetic variation at a few or sev- that genetic polymorphisms may play in several aspects eral loci that determine the uptake and metabolism of var- of human nutrition, and the ill-defined terms nutrige- ious nutrients. -
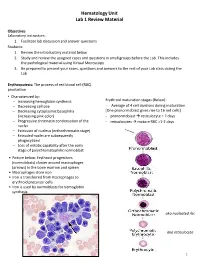
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

SEED Haematology Sysmex Educational Enhancement and Development October 2012
SEED Haematology Sysmex Educational Enhancement and Development October 2012 The red blood cell indices The full blood count has been used in conjunction with the traditional red The complete blood count (CBC) is central to clinical deci- cell indices in order to narrow down the possible causes sion making. This makes it one of the commonest laboratory of anaemia in an individual patient. investigations performed worldwide. Whilst the definition of what constitutes an CBC is influenced by the number Impedance technology and type of parameters measured by different haematology The RBC, HCT and MCV are all closely interrelated as they analysers, the traditional red cell indices that are widely are derived from information obtained from the passage used to classify anaemias are common to all. of cells through the aperture of the impedance channel of an automated haematology analyser. The impedance The laboratory approach to anaemia technology is based on the principle that an electrical field, Anaemia is an extremely common global healthcare prob- created between two electrodes of opposite charge, can lem. However, anaemia is merely a symptom which can be used to count and determine the size of cells. Blood result from a multitude of causes. Effective treatment is cells are poor conductors of electricity. The diluent in which only possible if the underlying cause is correctly identified. they are suspended as they pass through the aperture To this end, several classification systems have been devis- during counting is an isotonic solution which is a good ed. The most useful and widely used classification system conductor of electricity. -

Acoi Board Review 2019 Text
CHERYL KOVALSKI, DO FACOI NO DISCLOSURES ACOI BOARD REVIEW 2019 TEXT ANEMIA ‣ Hemoglobin <13 grams or ‣ Hematocrit<39% TEXT ANEMIA MCV RETICULOCYTE COUNT Corrected retic ct = hematocrit/45 x retic % (45 considered normal hematocrit) >2%: blood loss or hemolysis <2%: hypoproliferative process TEXT ANEMIA ‣ MICROCYTIC ‣ Obtain and interpret iron studies ‣ Serum iron ‣ Total iron binding capacity (TIBC) ‣ Transferrin saturation ‣ Ferritin-correlates with total iron stores ‣ can be normal or increased if co-existent inflammation TEXT IRON DEFICIENCY ‣ Most common nutritional problem in the world ‣ Absorbed in small bowel, enhanced by gastric acid ‣ Absorption inhibited by inflammation, phytates (bran) & tannins (tea) TEXT CAUSES OF IRON DEFICIENCY ‣ Blood loss – most common etiology ‣ Decreased intake ‣ Increased utilization-EPO therapy, chronic hemolysis ‣ Malabsorption – gastrectomy, sprue ‣ ‣ ‣ TEXT CLINICAL MANIFESTATIONS OF IRON DEFICIENCY ‣ Impaired psychomotor development ‣ Fatigue, Irritability ‣ PICA ‣ Koilonychiae, Glossitis, Angular stomatitis ‣ Dysphagia TEXT IRON DEFICIENCY LAB FINDINGS ‣ Low serum iron, increased TIBC ‣ % sat <20 TEXT MANAGEMENT OF IRON DEFICIENCY ‣ MUST LOOK FOR SOURCE OF BLEED: ie: GI, GU, Regular blood donor ‣ Replacement: 1. Oral: Ferrous sulfate 325 mg TID until serum iron, % sat, and ferritin mid-range normal, 6-12 months 2. IV TEXT SIDEROBLASTIC ANEMIAS Diverse group of disorders of RBC production characterized by: 1. Defect involving incorporation of iron into heme molecule 2. Ringed sideroblasts in