Morphologic Evaluation of Anemia – I Adewoyin S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oral Diagnosis: the Clinician's Guide
Wright An imprint of Elsevier Science Limited Robert Stevenson House, 1-3 Baxter's Place, Leith Walk, Edinburgh EH I 3AF First published :WOO Reprinted 2002. 238 7X69. fax: (+ 1) 215 238 2239, e-mail: [email protected]. You may also complete your request on-line via the Elsevier Science homepage (http://www.elsevier.com). by selecting'Customer Support' and then 'Obtaining Permissions·. British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Cataloging in Publication Data A catalog record for this book is available from the Library of Congress ISBN 0 7236 1040 I _ your source for books. journals and multimedia in the health sciences www.elsevierhealth.com Composition by Scribe Design, Gillingham, Kent Printed and bound in China Contents Preface vii Acknowledgements ix 1 The challenge of diagnosis 1 2 The history 4 3 Examination 11 4 Diagnostic tests 33 5 Pain of dental origin 71 6 Pain of non-dental origin 99 7 Trauma 124 8 Infection 140 9 Cysts 160 10 Ulcers 185 11 White patches 210 12 Bumps, lumps and swellings 226 13 Oral changes in systemic disease 263 14 Oral consequences of medication 290 Index 299 Preface The foundation of any form of successful treatment is accurate diagnosis. Though scientifically based, dentistry is also an art. This is evident in the provision of operative dental care and also in the diagnosis of oral and dental diseases. While diagnostic skills will be developed and enhanced by experience, it is essential that every prospective dentist is taught how to develop a structured and comprehensive approach to oral diagnosis. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

Molecular Basis of Spectrin Deficiency in Beta Spectrin Durham. a Deletion
Molecular basis of spectrin deficiency in beta spectrin Durham. A deletion within beta spectrin adjacent to the ankyrin-binding site precludes spectrin attachment to the membrane in hereditary spherocytosis. H Hassoun, … , S S Chiou, J Palek J Clin Invest. 1995;96(6):2623-2629. https://doi.org/10.1172/JCI118327. Research Article We describe a spectrin variant characterized by a truncated beta chain and associated with hereditary spherocytosis. The clinical phenotype consists of a moderate hemolytic anemia with striking spherocytosis and mild spiculation of the red cells. We describe the biochemical characteristics of this truncated protein which constitutes only 10% of the total beta spectrin present on the membrane, resulting in spectrin deficiency. Analysis of reticulocyte cDNA revealed the deletion of exons 22 and 23. We show, using Southern blot analysis, that this truncation results from a 4.6-kb genomic deletion. To elucidate the basis for the decreased amount of the truncated protein on the membrane and the overall spectrin deficiency, we show that (a) the mutated gene is efficiently transcribed and its mRNA abundant in reticulocytes, (b) the mutant protein is normally synthesized in erythroid progenitor cells, (c) the stability of the mutant protein in the cytoplasm of erythroblasts parallels that of the normal beta spectrin, and (d) the abnormal protein is inefficiently incorporated into the membrane of erythroblasts. We conclude that the truncation within the beta spectrin leads to inefficient incorporation of the mutant protein into the skeleton despite its normal synthesis and stability. We postulate that this misincorporation results from conformational changes of the beta spectrin subunit affecting the binding of the abnormal heterodimer to ankyrin, and we provide evidence […] Find the latest version: https://jci.me/118327/pdf Molecular Basis of Spectrin Deficiency in p8 Spectrin Durham A Deletion within .3 Spectrin Adjacent to the Ankyrin-binding Site Precludes Spectrin Attachment to the Membrane in Hereditary Spherocytosis Hani Hassoun,* John N. -

Erythrocytes: Overview, Morphology, Quantity by AH Rebar Et
In: A Guide to Hematology in Dogs and Cats, Rebar A.H., MacWilliams P.S., Feldman B.F., Metzger F.L., Pollock R.V.H. and Roche J. (Eds.). Publisher: Teton NewMedia, Jackson WY (www.veterinarywire.com). Internet Publisher: International Veterinary Information Service, Ithaca NY (www.ivis.org), 8-Feb-2005; A3304.0205 Erythrocytes: Overview, Morphology, Quantity A.H. Rebar1, P.S. MacWilliams2, B.F. Feldman 3, F.L. Metzger 4, R.V.H. Pollock 5 and J. Roche 6 1Dept of Veterinary Pathobiology, School of Veterinary Medicine, Purdue University, IN,USA. 2Dept of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, WI, USA. 3Dept of Biomedical Sciences & Pathobiology, VA-MD - Regional College of Veterinary Medicine, Virginia Tech, VA, USA. 4Metzger Animal Hospital,State College,PA, USA. 5Fort Hill Company, Montchanin, DE, USA. 6 Hematology Systems, IDEXX Laboratories, Westbrook, ME, USA. Overview Production Red blood cells (RBC) are produced in the bone marrow. Numbers of circulating RBCs are affected by changes in plasma volume, rate of RBC destruction or loss, splenic contraction, erythropoietin (EPO) secretion, and the rate of bone marrow production. A normal PCV is maintained by an endocrine loop that involves generation and release of erythropoietin (EPO) from the kidney in response to renal hypoxia. Erythropoietin stimulates platelet production as well as red cell production. However, erythropoietin does not stimulate white blood cell (WBC) production. Erythropoiesis and RBC numbers are also affected by hormones from the adrenal cortex, thyroid, ovary, testis, and anterior pituitary. Destruction Red cells have a finite circulating lifespan. In dogs, the average normal red cell circulates approximately 100 days. -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

We Get Biased on Things That Make Sense
DIALOGUE AND DISCUSSION We Get Biased on Things That Make Sense MERIH T. TESFAZGHI When I was teaching third year medical students, and bench- reaction might be characterized by a marked left shift lecturing clinical laboratory science students, I emphasized which may display a range of immature cells that are also the importance of clinical details and the history of patients seen in CML. Severe left shift especially in the absence of in laboratory test processing, especially in the evaluation of evident infection (or other underlying diseases) may peripheral blood films. During those times, one of my strongly suggest direct bone marrow involvement. In students stated, “but including details of patients might such scenario, clinical details of patients such as absence somehow affect the decision of the reviewer, and may lead to or presence of enlargement of extramedullar organs is Downloaded from a bias.” I responded that we are biased on things that make highly sought. sense. This means that we decide based on the evidence we see from a microscopy evaluation in the light of the clinical Ø The coexistence of multiple conditions in a given detail and history. How do clinical details and history help us patient. One condition may mask the presence of the reach decisions? other condition. For example, the coexistence of megaloblastic anemia with iron deficiency anemia. http://hwmaint.clsjournal.ascls.org/ If specimens are the “in vitro ambassadors” of patients to the Usually, megaloblastic anemia is characterized by laboratory, then properly completed request forms are their increased size of red blood cells (MCV), but when “credentials”. -
![Anormal Rbc in Peripheral Blood. [Repaired].Pdf](https://docslib.b-cdn.net/cover/4277/anormal-rbc-in-peripheral-blood-repaired-pdf-544277.webp)
Anormal Rbc in Peripheral Blood. [Repaired].Pdf
1. Acanthocyte 2. Burr-cell 3. Microcyte 1. Basophilic Normoblast 2. Polychromatic Normoblast 3. Pycnotic Normoblast 4. Plasmocyte 5. Eosinophil 6. Promyelocyte 1. Macrocyte 2. Elliptocyte 1. Microcyte 2. Normocyte 1. Polychromatic Erythrocyte 2. Acanthocyte 3. Elliptocyte 1. Polychromatic Normoblast 2. Pycnotic Normoblast 3. Neutrophil Myelocyte 4. Neutrophil Metamyelocyte 1. Schistocyte 2. Microcyte BASOPHILIC ( EARLY ) NORMOBLASTS Basophilic Erythroblast Basophilic Stippling, Blood smear, May-Giemsa stain, (×1000) CABOT'S RINGS Drepanocyte Elliptocyte Erythroblast ERYTHROBLAST in the blood Howell-jolly body Hypo chromic LACRYMOCYTES Leptocyte Malaria, Blood smear, May-Giemsa stain, ×1000 MICROCYTES Orthochromatic erythroblast Pappen heimer Bodies & 1. Schistocyte 2. Elliptocyte 3. Acanthocyte POIKILOCYTOSIS Polychromatic Erythroblast Pro Erytroblast Proerythroblasts Reticulocyte Rouleaux SICKLE CELLS Sickle cell Spherocyte Spherocyte Spherocyte SPHEROCYTES STOMATOCYTES Target Cells Tear Drop Cell, Blood smear, May-Giemsa stain, x1000 Anulocyte 1. Burr-cell 2. Elliptocyte 1. Macrocyte 2. Microcyte 3. Elliptocyte 4. Schistocyte 1. Ovalocyte 2. Lacrymocyte 3. Target cell 1. Polychromatic Erythrocyte 2. Basophilic Stippling 1. Proerythroblast 2. Basophilic Erythroblast 3. Intermediate Erythroblast 4. Late Erythroblast 5. Monocyte 6. Lymphocyte 1. Target-cell 2. Elliptocyte 3. Acanthocyte 4. Stomatocyte 5. Schistocyte 6. Polychromatophilic erythrocyte. 1.Pro erythroblast 2.Basophilic normoblast 3.Polychromatic normoblast 4.Pycnotic normoblast -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets. -
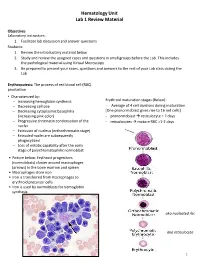
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Rbcs) in Both the Sinus and Cord, Histiocytes Werc Swollen by a Granu}Ar Substance in the Cytoplasm and Also Many Secondary Lysosomes
The UOEHAssociationUOEH Association ofofHealth Health Sciences JUOEH20(1)!11-19 (1998) 11 [Original) Pathological Study Splenomegaly Associated with Cadmium-inducedof Anemia in Rats Tetsuo HAMADA', Akihide TANIMOT02, Nobuyuki ARIMA!, Yoshihiro IDEL, Takakazu SASAGURI2, Shohei SHIMiVIRI2, Yoshitaka MURATA', Ke-Yong WANG2 and Yasuyuki SASAGURIL' 'Dopartment of'Surgical Pathotogy, University llosPital ?Dopartment qf Palhology and Cell Biotogy, Scheol of' Adlrdicine, (1itib'ersity of Occapational and Environmental Health., .lapan. }'dhatanishi-ku, Kiialp,ztshu 807-8imr, .1apan Ahstract : Splenomegaly was observcd both in male and R)malc Spraguc-Dawley rats after 1 week of exposure to CdC12 (O,6 mg (:d/kg/day), Sp]een weight reached about double that in controls by 8 weeks of Cd exposure. Histopathologica] cxamination of the enlargcd spleen rcvcaled that iron- and lipid-laden histiocytes were clustered in tha periarterial lymphatic sheath, and the red pulp appeared to be expanded. It is noteworthy that electron microscopy rcvealed markcd poikilocytosis and Hcinz body formation in red blood cells (RBCs) in both the sinus and cord, Histiocytes werc swollen by a granu}ar substance in the cytoplasm and also many secondary lysosomes. Thesc morphological findings indicatc that degradation of damagcd RBCs induced by exposure to Cd might bc promoted in the splccn and possibly cause splenomcgaly, This RBC damage-hcmolysis-splcnomegaly sequence is also considered to be associated with the etioiogy of Cd-induced anemia, In addition to the abnormal RBC degradation, nuclel of ]ymphocytes in thc Cd-cxposcd spleen exhibited high elcctron density, consistent with a preapoptotic statc suggesting the immunosupprcssive effhct ofCd. Kay woralf :spienomegaly, poiki]ocytosis, Heinz body, cadmiurn, anemia, (Received 18 November 1997, accepted I9January 1998) Introduction Cd was reported to cause anemia in dogs as early as 1896 [1], and subsequently, anemia was found in humans after Cd exposure [2]. -

Copyrighted Material
CHAPTER 1 The Blood Film and Count Blood Blood is a life‐sustaining fluid that circulates through the heart and blood vessels. It carries oxygen and nutrients to the tissues and waste products to the lungs, liver and kidneys, where they can be removed from the body. Usually when blood is removed from the body it forms a solid blood clot. However, if clotting is prevented by mixing with an anticoagulant, the blood separates, under the influence of gravity, into three layers (Fig. 1.1). The bottom layer is deep red in colour and is composed of red cells. The top layer is clear and pale yellow. It is called plasma and is composed of various salts and proteins dissolved in water. In between is a narrow layer called the buffy coat because of its buff or yellowish white colour. The buffy coat is composed mainly of cells of a variety of types, collectively known as white cells. In addition there are small cellular fragments, called platelets, which have a role in blood clotting. The bloodCOPYRIGHTED film MATERIAL Although we can judge the proportions of red cells and white cells in a tube of sedimented blood, we get far more information if the blood is carefully mixed and a thin layer is spread on a glass A Beginner’s Guide to Blood Cells, Third Edition. Barbara J. Bain. © 2017 John Wiley & Sons Ltd. Published 2017 by John Wiley & Sons Ltd. 1 0003049575.indd 1 2/24/2017 9:50:31 AM 2 A Beginner’s Guide to Blood Cells Plasma Buffy coat Red cells Fig. -

The Clinical Application of Della-Porta Et Al Score in a Mexican Patient with Myelodysplastic Syndrome
CLINICAL CASE Rev Hematol Mex. 2020; 21 (3): 158-171. The clinical application of Della-Porta et al score in a Mexican patient with myelodysplastic syndrome. Aplicación clínica de la puntuación Della-Porta y colaboradores en un paciente mexicano con síndrome mielodisplásico Juan C Marín-Corte,1,4 Eduardo Olmedo-Gutiérrez,1,4 Jeny A Marín-Corte,5 Roberto N Miranda,3 Carlos E Bueso-Ramos,3 Omar Cano-Jiménez,1 Arturo R Fuentes-Reyes,1 Elizabeth Hernández-Salamanca,1,4 Miguel A López-Trujillo,1,4 Rafael A Marín-López,1,4 Guillermo J Ruiz-Delgado,1,2,4 Guillermo J Ruiz-Argüelles1,2,4 Abstract BACKGROUND: The myelodysplastic syndromes are one of the most studied diseases in hematology in recent years. By definition and according to the World Health Or- ganization Classification of Tumours of hematopoietic and Lymphoid Tissues 2017, myelodysplastic syndromes are a group of clonal hematopoietic stem cell diseases characterized by cytopenia, dysplasia in one or more of the major myeloid lineages, ineffective hematopoiesis, recurrent genetic abnormalities and increased risk of de- veloping acute myeloid leukemia (AML). To identify in a quick way this disease, we designed a worksheet based on the article of Matteo Della-Porta et al to developed a systematic approach to assess the morphological features in the bone marrow smears of three cell lineages in patients with myelodysplastic syndromes and provide the basis to validate flow cytometric and immunohistochemistry data. CLINICAL CASE: A 47-year-old male patient in whom we applied a worksheet that we designed based on Della-Porta score criteria to each cellularity lineage in the bone 1 Clinical Laboratories of Puebla; Puebla, marrow smears and we obtained significant results according to the bone marrow Puebla, México.