Brafnon-V600E More Frequently Co-Occurs with IDH1/2 Mutation in Adult Patients with Gliomas Than Patients Harboring BRAFV600E, but Without Survival Advantage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Influencers on Thyroid Cancer Onset: Molecular Genetic Basis
G C A T T A C G G C A T genes Review Influencers on Thyroid Cancer Onset: Molecular Genetic Basis Berta Luzón-Toro 1,2, Raquel María Fernández 1,2, Leticia Villalba-Benito 1,2, Ana Torroglosa 1,2, Guillermo Antiñolo 1,2 and Salud Borrego 1,2,* 1 Department of Maternofetal Medicine, Genetics and Reproduction, Institute of Biomedicine of Seville (IBIS), University Hospital Virgen del Rocío/CSIC/University of Seville, 41013 Seville, Spain; [email protected] (B.L.-T.); [email protected] (R.M.F.); [email protected] (L.V.-B.); [email protected] (A.T.); [email protected] (G.A.) 2 Centre for Biomedical Network Research on Rare Diseases (CIBERER), 41013 Seville, Spain * Correspondence: [email protected]; Tel.: +34-955-012641 Received: 3 September 2019; Accepted: 6 November 2019; Published: 8 November 2019 Abstract: Thyroid cancer, a cancerous tumor or growth located within the thyroid gland, is the most common endocrine cancer. It is one of the few cancers whereby incidence rates have increased in recent years. It occurs in all age groups, from children through to seniors. Most studies are focused on dissecting its genetic basis, since our current knowledge of the genetic background of the different forms of thyroid cancer is far from complete, which poses a challenge for diagnosis and prognosis of the disease. In this review, we describe prevailing advances and update our understanding of the molecular genetics of thyroid cancer, focusing on the main genes related with the pathology, including the different noncoding RNAs associated with the disease. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Epigenetic Loss of the RNA Decapping Enzyme NUDT16 Mediates C-MYC Activation in T-Cell Acute Lymphoblastic Leukemia
OPEN Leukemia (2017) 31, 1622–1657 www.nature.com/leu LETTERS TO THE EDITOR Epigenetic loss of the RNA decapping enzyme NUDT16 mediates C-MYC activation in T-cell acute lymphoblastic leukemia Leukemia (2017) 31, 1622–1625; doi:10.1038/leu.2017.99 Having found the aforementioned NUDT16 CpG island methy- lation profiles, we studied in greater detail their association with the possible transcriptional inactivation of the NUDT16 gene at the RNA and protein levels in leukemia cell lines. We first It is possible that the occurrence of intrinsic defects in RNA performed bisulfite genomic sequencing of mutiple clones in the – processing pathways, such as RNA decapping,1 3 contribute to the T-cell Acute Lymphoblastic Leukemia (T-ALL) cell lines CCRF-CEM, distorted RNA landscapes of cancer cells. After transcription by Jurkat, MOLT-4 and MOLT-16 using primers that encompassed the RNA polymerase II, RNA molecules are equipped with a 5´-end transcription start site-associated CpG island and confirmed the N7-methyl guanosine (m7G)-cap. This m7G-cap is essential for hypermethylated status of the 5′-end region of NUDT16 in translation, stabilizing the RNA molecule and protecting it from comparison to normal T lymphocytes (Figure 1b), validating the – exonucleolytic breakdown.1 3 For RNA decay to occur the DNA methylation patterns obtained by the microarray approach m7G-cap first needs to be removed. This process is known as (Supplementary Figure S2). In contrast, normal T lymphocytes, the – decapping.1 3 The decapping mRNA 2 (DCP2) enzyme,4 also T-ALL cell lines KOPN-8, REH and RS4;11 and the leukemia cell known as the nucleoside diphosphate-linked moiety X motif lines HL-60 and K562 derived from myeloid lineage were all found 20 (NUDT20), was originally thought to be the only mammalian to be unmethylated (Figure 1b; Supplementary Figure S2). -
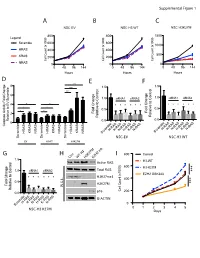
C a B D G E F
Supplemental Figure 1 A B C NSC EV NSC H3 WT NSC H3K27M 800 800 1500 ) ) Legend ) Scramble 600 600 x1000 x1000 x1000 ( ( ( 1000 t t t n n HRAS n 400 400 u u u o o o C C C 500 l l KRAS l 200 200 el el el C C C NRAS 0 0 0 0 48 96 144 0 48 96 144 0 48 96 144 Hours Hours Hours D *** E F 8 1.5 1.5 e *** l l *** ro ro t 6 t 1.0 siRNA1 siRNA2 1.0 siRNA1 siRNA2 Con Con o o t 4 * * t * * * * * * e * * * * * * e v i * * v i t 0.5 * * t 0.5 Fold Change 2 Fold Change Rela Rela Relative to EV Scrambl 0.0 0.0 0 Caspase Activity Fold Change H-RASK-RASN-RASH-RASK-RASN-RAS H-RASK-RASN-RASH-RASK-RASN-RAS KRAS KRAS KRAS HRAS NRAS HRAS NRAS HRAS NRAS Scramble Scramble Scramble Scramble Scramble NSC-EV NSC-H3 WT EV H3WT H3K27M G H I 800 Control Con WT H3 H3K27MEZH2 inb 1.5 l H3-WT Active RAS ro ) t 600 H3-K27M 1.0 siRNA1 siRNA2 Total RAS *** Con x1000 ( o EZH2 GSK343 t t * * * * * * H3K27me3 n e 400 u v i o t 0.5 *** H3K27M C Fold Change l W.C.L. el Rela p16 C 200 0.0 B-ACTIN H-RASK-RASN-RASH-RASK-RASN-RAS 0 Scramble NSC-H3 K27M 0 1 2 3 4 5 Days E C A 100 Cell Viability 100 25 50 75 Cell Viability 25 50 75 0 0 Fold Change MOCK MOCK Relative to Control Scramble * 0.0 0.2 0.4 0.6 0.8 1.0 1.2 MYC Scramble MYC MYC * PDGFRA Scramble siRNA PDGFRA MYC PDGFRA AURKA AURKA DIPG007siRNA2 PDGFRA NSC H3K27MsiRNA1and AURKA LAMTOR3 AURKA LAMTOR3 LAMTOR3 LIN28A LAMTOR3 LIN28A LIN28B LIN28A NSC-EV LIN28A MAP2K3 LIN28B LIN28B LIN28B MAP2K5 siRNA1 MAP2K3 MAP2K3 MAP3K2 MAP2K3 MAP3K7 MAP2K5 MAP2K5 MAPK7 MAP2K5 2 MAP3K2 siRNA1 siRNA2 MAP3K2 ZAK MAP3K7 MAP3K2 MAP3K7 MAPK7 MAP3K7 -

Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in Vitro and in Vivo
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Genetik Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in vitro and in vivo - Towards Response Prediction in Cancer Therapy with Kinase Inhibitors Michaela Bairlein Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ. -Prof. Dr. K. Schneitz Prüfer der Dissertation: 1. Univ.-Prof. Dr. A. Gierl 2. Hon.-Prof. Dr. h.c. A. Ullrich (Eberhard-Karls-Universität Tübingen) 3. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 07.01.2010 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 19.04.2010 angenommen. FOR MY PARENTS 1 Contents 2 Summary ................................................................................................................................................................... 5 3 Zusammenfassung .................................................................................................................................................... 6 4 Introduction .............................................................................................................................................................. 8 4.1 Cancer .............................................................................................................................................................. -

MAP2K4/MKK4 Expression in Pancreatic Cancer: Genetic Validation of Immunohistochemistry and Relationship to Disease Course
8516 Vol. 10, 8516–8520, December 15, 2004 Clinical Cancer Research MAP2K4/MKK4 Expression in Pancreatic Cancer: Genetic Validation of Immunohistochemistry and Relationship to Disease Course Wei Xin,1 Ki J. Yun,4 Francesca Ricci,2 ing patterns were also evaluated among unresectable pri- Marianna Zahurak,3 Wanglong Qiu,4 mary and metastatic cancer tissues from autopsy specimens, Gloria H. Su,4 Charles J. Yeo,5,6 indicating intact Mkk4 immunolabeling in 88.8% of the unresectable primary carcinomas as compared with 63.3% Ralph H. Hruban,4,5 Scott E. Kern,4,5 and 4,5 of distant metastases (P < 0.001). Our data indicate that the Christine A. Iacobuzio-Donahue loss of Mkk4 protein expression in pancreatic carcinomas 1 Department of Pathology, The University of Michigan Medical may be more frequent than suggested by the rates of genetic Center, Ann Arbor, Michigan; 2Department of Pathology, University La Sapienza, Rome, Italy; and the Departments of 3Biostatistics, inactivation alone and that MKK4 loss may contribute to 4Pathology, 5Oncology, and 6Surgery, The Johns Hopkins University disease progression. The correlation of MKK4 genetic status Hospital, Baltimore, Maryland with immunolabeling patterns validate this approach for the evaluation of MKK4 status in routine histologic sections and ABSTRACT may provide useful information regarding patient prognosis. MKK4 (MAP2K4/SEK1) is a member of the mitogen- activated protein kinase family, originally identified as a INTRODUCTION kinase involved in the stress-activated protein kinase path- The mitogen-activated protein (MAP) kinase cascades are way by directly phosphorylating c-Jun NH -terminal kinase. 2 multifunctional signaling pathways that are evolutionally well MKK4 genetic inactivation has been observed in a subset of conserved in all of the eukaryotic cells. -

Molecular Mechanisms Underlying Innate Immune Kinase
MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION APPROVED BY SUPERVISORY COMMITTEE Michael A White, Ph.D. Melanie H. Cobb, Ph.D. Lawrence Lum, Ph.D. John D. Minna, M.D. DEDICATION This work is dedicated to my mother and Arlene for their love and support. ACKNOWLEDGEMENTS I am very grateful to my mentor, Dr. Michael White, for his continuous support and guidance through the entire study. I really appreciate his inspiration, patience, and generosity. I would also like to thank my committee members, Dr. Cobb, Dr. Lum, and Dr. Minna, for their invaluable advice and discussion. I thank all the White lab members and my friends for their help, suggestion, and discussion. Particularly, I would like to thank Rosie, Michael, Brian, Tzuling, and Malia for their long-term support and collaboration. I would also like to thank my friends, Veleka, Pei-Ling, Jen-Chieh, Shu-Yi, Chih-Chiang, Jen-Shuan, Yi-Chun, and Yu-San for their friendship. I am grateful to Drs. Rolf Brekken, Zhijian James Chen, Xuetao Cao, Philip Tsichlis, Charles Yeaman, William Hahn, Keqiang Ye, and Shu-Chan Hsu, Bing Su, Dos Sarbassov, Mark Magnuson, David Sabatini, Thomas Tan, and Bert Vogelstein for many of the reagents used in these studies. Finally, and most importantly, I would like to thank my mother and Arlene for their unending support and encouragement. MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION by YI-HUNG OU DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas April, 2013 Copyright by YI-HUNG OU, 2013 All Rights Reserved MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION Publication No. -
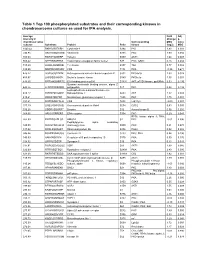
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S
crossmark Research Author’s Choice © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S Rolf Turk‡§¶ʈ**, Jordy J. Hsiao¶, Melinda M. Smits¶, Brandon H. Ng¶, Tyler C. Pospisil‡§¶ʈ**, Kayla S. Jones‡§¶ʈ**, Kevin P. Campbell‡§¶ʈ**, and Michael E. Wright¶‡‡ Mutations in genes encoding components of the sar- The muscular dystrophies are hereditary diseases charac- colemmal dystrophin-glycoprotein complex (DGC) are re- terized primarily by the progressive degeneration and weak- sponsible for a large number of muscular dystrophies. As ness of skeletal muscle. Most are caused by deficiencies in such, molecular dissection of the DGC is expected to both proteins associated with the cell membrane (i.e. the sarco- reveal pathological mechanisms, and provides a biologi- lemma in skeletal muscle), and typical features include insta- cal framework for validating new DGC components. Es- bility of the sarcolemma and consequent death of the myofi- tablishment of the molecular composition of plasma- ber (1). membrane protein complexes has been hampered by a One class of muscular dystrophies is caused by mutations lack of suitable biochemical approaches. Here we present in genes that encode components of the sarcolemmal dys- an analytical workflow based upon the principles of pro- tein correlation profiling that has enabled us to model the trophin-glycoprotein complex (DGC). In differentiated skeletal molecular composition of the DGC in mouse skeletal mus- muscle, this structure links the extracellular matrix to the cle. We also report our analysis of protein complexes in intracellular cytoskeleton. -

Members of the Competence Network for Congenital Heart Defects, Germany
Members of the Competence Network for Congenital Heart Defects, Germany Hashim Abdul-Khaliq, Hans-Heiner Kramer, Felix Berger, Brigitte Stiller, Ulrike Bauer, Thomas Pickardt, Sabine Klaassen Family 49 Family 62 Family 226 Family 333 * * * * * * * * TOF AVS ASD ASD PAPVR TOF * * * * ASD PAPVD COA ASD * Family 346 Family 398 VSD * Family 489 * * VSD VSD * * VSD HCM * * * Family 545 BAV AVS ASD PDA Sv AS VSD Family 576 Family 645 Septal defect * * * COA AVS ASD BAV VSD * * * * BAV AVS AVS ASD BAV Family 702 * VSD VSD Family 732 * Family 720 AVSD * * * VSD * * Family 831 TOF TOF TOF Vring TGA VSD * * * * PDA VSD AVS * PDA * * * VSD BAV PFO ASD ASD2 PDA PDA COA Family 1117 Family 1121 * PVS ? * ASD VSD * * * TOF VSD ASD VSD Family 1319 Family 1151 * * * * 2 * ASD ASD * * ASD ASD * * EbA AVS AVS Family 1364 Family 1560 Family 1575 * * TOF VSD ASD VSD infPS TOF * * * VSD PVS PVS PVS VSD * VSD PVS Family 1710 ASD Family 1722 * ASD CHD * VSD VSD * COA TGA DCM ASD BAV SV, MVA COA PVS * DCM DCM ASD HLHS COA BAV Family 2077 Family 2261 * COA, BAV * * * PVS PVS PVS * * infPS infPS BAV ASD * * AVS ASD ASD BAV BAV Family 3500 Family 2558 Family 3315 * * * AVS VSD AVR * * HLHS COA PDA ASD BAV * PVA Family 3501 VSD Family 3503 ? ? * * * VSD HRHS EbA * 3 PVA PAA ASD PDA VSD Family 3505 Family 3540 * * BAV ASD AVS * * * HLHS HLHS TAPVR BAV COA Figure S1. Pedigrees of 32 Danish multiplex CHD families. Circles: females. Squares: males. White symbols: unaffected family members. Filled symbols: affected family members. Triangles: abortion. -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA.