Molecular Mechanisms Underlying Innate Immune Kinase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Influencers on Thyroid Cancer Onset: Molecular Genetic Basis
G C A T T A C G G C A T genes Review Influencers on Thyroid Cancer Onset: Molecular Genetic Basis Berta Luzón-Toro 1,2, Raquel María Fernández 1,2, Leticia Villalba-Benito 1,2, Ana Torroglosa 1,2, Guillermo Antiñolo 1,2 and Salud Borrego 1,2,* 1 Department of Maternofetal Medicine, Genetics and Reproduction, Institute of Biomedicine of Seville (IBIS), University Hospital Virgen del Rocío/CSIC/University of Seville, 41013 Seville, Spain; [email protected] (B.L.-T.); [email protected] (R.M.F.); [email protected] (L.V.-B.); [email protected] (A.T.); [email protected] (G.A.) 2 Centre for Biomedical Network Research on Rare Diseases (CIBERER), 41013 Seville, Spain * Correspondence: [email protected]; Tel.: +34-955-012641 Received: 3 September 2019; Accepted: 6 November 2019; Published: 8 November 2019 Abstract: Thyroid cancer, a cancerous tumor or growth located within the thyroid gland, is the most common endocrine cancer. It is one of the few cancers whereby incidence rates have increased in recent years. It occurs in all age groups, from children through to seniors. Most studies are focused on dissecting its genetic basis, since our current knowledge of the genetic background of the different forms of thyroid cancer is far from complete, which poses a challenge for diagnosis and prognosis of the disease. In this review, we describe prevailing advances and update our understanding of the molecular genetics of thyroid cancer, focusing on the main genes related with the pathology, including the different noncoding RNAs associated with the disease. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Targeting the Hippo Pathway in Prostate Cancer: What's New?
cancers Review Targeting the Hippo Pathway in Prostate Cancer: What’s New? Kelly Coffey Solid Tumour Target Discovery Laboratory, Translational and Clinical Research Institute, Newcastle University Centre for Cancer, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK; [email protected] Simple Summary: Prostate cancer is the most commonly diagnosed cancer in men in the UK, accounting for the deaths of over 11,000 men per year. A major problem in this disease are tumours which no longer respond to available treatments. Understanding how this occurs will reveal new ways to treat these patients. In this review, the latest findings regarding a particular group of cellular factors which make up a signalling network called the Hippo pathway will be described. Accumulating evidence suggests that this network contributes to prostate cancer progression and resistance to current treatments. Identifying how this pathway can be targeted with drugs is a promising area of research to improve the treatment of prostate cancer. Abstract: Identifying novel therapeutic targets for the treatment of prostate cancer (PC) remains a key area of research. With the emergence of resistance to androgen receptor (AR)-targeting therapies, other signalling pathways which crosstalk with AR signalling are important. Over recent years, evidence has accumulated for targeting the Hippo signalling pathway. Discovered in Drosophila melanogasta, the Hippo pathway plays a role in the regulation of organ size, proliferation, migration and invasion. In response to a variety of stimuli, including cell–cell contact, nutrients and stress, a kinase cascade is activated, which includes STK4/3 and LATS1/2 to inhibit the effector proteins YAP and its paralogue TAZ. -

MRT67307 Kinase Inhibitor; TBK1 and Ikkε Inhibitor Catalog Code: Inh-Mrt for Research Use Only Version 19E07-NJ
MRT67307 Kinase inhibitor; TBK1 and IKKε inhibitor Catalog Code: inh-mrt https://www.invivogen.com/mrt67307 For research use only Version 19E07-NJ PRODUCT INFORMATION CHEMICAL PROPERTIES Contents CAS Number: 1190378-57-4 (free base) • 10 mg MRT67307 (hydrochloride) Formula: C26H36N6O2 . x HCl Molecular weight: 464.60 g/mol (free base) Storage and stability Solubility: 15 mg/ml H2O - MRT67307 is provided as a dried powder and shipped at room temperature. Upon receipt, store product at -20 °C. METHODS - Upon resuspension of MRT67307 prepare aliquots and store Preparation of stock solution (10 mg/ml) at -20 °C. Resuspended product is stable for at least 3 months when 1. Add 1ml of endotoxin-free H O properly stored. 2 2. Use immediately or store aliquots at -20 °C - Avoid repeated freeze-thaw cycles. 3. Prepare dilutions using sterile endotoxin-free water Quality control Working concentration range: 1 - 20 µM (for cell culture assays) - Purity: ≥95% (UHPLC) - Inhibition of TBK1/IKKε by MRT67307 has been confirmed using Inhibition of TBK1/IKKε by MRT67307 in a cellular assay cellular assays. Below is a protocol using InvivoGen’s THP1-Dual™ cells for studying - Absence of bacterial contamination (e.g. lipoproteins and endotoxins) specific inhibition of the IRF pathway by MRT67307. These cells has been confirmed using HEK-Blue™ hTLR2 and HEK-Blue™ hTLR4 cells. express both an inducible Lucia luciferase reporter and an inducible secreted embryonic alkaline phosphatase (SEAP) reporter to measure PRODUCT DESCRIPTION the activation of the IRF or NF-κB pathways, respectively. Changes in MRT67307 is a potent, reversible kinase inhibitor, and a derivative the Lucia expression levels upon inhibition can be readily assessed by of BX7951. -

Mechanisms of IKBKE Activation in Cancer Sridevi Challa University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School 1-29-2017 Mechanisms of IKBKE Activation in Cancer Sridevi Challa University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Biochemistry Commons, Biology Commons, and the Cell Biology Commons Scholar Commons Citation Challa, Sridevi, "Mechanisms of IKBKE Activation in Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6617 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Mechanisms of IKBKE Activation in Cancer by Sridevi Challa A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Cell Biology, Microbiology, and Molecular Biology College of Arts and Sciences University of South Florida Major Professor: Mokenge P. Malafa, M.D. Gary Reuther, Ph.D. Eric Lau, Ph.D. Domenico Coppola, M.D. Date of Approval: January 12, 2017 Keywords: EGFR, Olaparib, resistance Copyright © 2017, Sridevi Challa DEDICATION This dissertation is dedicated to my kind and courageous mother. ACKNOWLEDGMENTS I would like to acknowledge Dr. Cheng for trusting me with completion of the projects. I would like to thank him for giving me the freedom to explore any aspect of research and always willing to provide the necessary resources and guidance for my projects. I want to also acknowledge Ted and the Cheng lab personnel for their support. -

Cep-2020-00633.Pdf
Clin Exp Pediatr Vol. 64, No. 5, 208–222, 2021 Review article CEP https://doi.org/10.3345/cep.2020.00633 Understanding the genetics of systemic lupus erythematosus using Bayesian statistics and gene network analysis Seoung Wan Nam, MD, PhD1,*, Kwang Seob Lee, MD2,*, Jae Won Yang, MD, PhD3,*, Younhee Ko, PhD4, Michael Eisenhut, MD, FRCP, FRCPCH, DTM&H5, Keum Hwa Lee, MD, MS6,7,8, Jae Il Shin, MD, PhD6,7,8, Andreas Kronbichler, MD, PhD9 1Department of Rheumatology, Wonju Severance Christian Hospital, Yonsei University Wonju College of Medicine, Wonju, Korea; 2Severance Hospital, Yonsei University College of Medicine, Seoul, Korea; 3Department of Nephrology, Yonsei University Wonju College of Medicine, Wonju, Korea; 4Division of Biomedical Engineering, Hankuk University of Foreign Studies, Yongin, Korea; 5Department of Pediatrics, Luton & Dunstable University Hospital NHS Foundation Trust, Luton, UK; 6Department of Pediatrics, Yonsei University College of Medicine, Seoul, Korea; 7Division of Pediatric Nephrology, Severance Children’s Hospital, Seoul, Korea; 8Institute of Kidney Disease Research, Yonsei University College of Medicine, Seoul, Korea; 9Department of Internal Medicine IV (Nephrology and Hypertension), Medical University Innsbruck, Innsbruck, Austria 1,3) The publication of genetic epidemiology meta-analyses has analyses have redundant duplicate topics and many errors. increased rapidly, but it has been suggested that many of the Although there has been an impressive increase in meta-analyses statistically significant results are false positive. In addition, from China, particularly those on genetic associa tions, most most such meta-analyses have been redundant, duplicate, and claimed candidate gene associations are likely false-positives, erroneous, leading to research waste. In addition, since most suggesting an urgent global need to incorporate genome-wide claimed candidate gene associations were false-positives, cor- data and state-of-the art statistical inferences to avoid a flood of rectly interpreting the published results is important. -

Iκb Kinase Ε and TANK-Binding Kinase 1 Activate AKT by Direct Phosphorylation
IκB kinase ε and TANK-binding kinase 1 activate AKT by direct phosphorylation Xiaoduo Xiea, Denghong Zhangb, Bin Zhaoa, Min-Kan Lua, Ming Youc, Gianluigi Condorellib, Cun-Yu Wangd, and Kun-Liang Guana,1 aDepartment of Pharmacology and Moores Cancer Center and bDepartment of Medicine, University of California at San Diego, La Jolla, CA 92093; cCancer Center, Medical College of Wisconsin, Milwaukee, WI 53226; and dLaboratory of Molecular Signaling, Division of Oral Biology and Medicine, University of California Los Angeles School of Dentistry, Los Angeles, CA 90095 Edited* by Jack E. Dixon, The Howard Hughes Medical Institute, Chevy Chase, MD, and approved March 8, 2011 (received for review October 27, 2010) AKT activation requires phosphorylation of the activation loop The noncanonical IκB kinase ε (IKKε) has been demonstrated (T308) by 3-phosphoinositide-dependent protein kinase 1 (PDK1) to play an essential role in Ras-induced cell transformation, which and the hydrophobic motif (S473) by the mammalian target of requires activation of both the Raf-MEK-ERK and PI3K-AKT rapamycin complex 2 (mTORC2). We recently observed that pathways (20). Of particular interest is the observation that IKKε phosphorylation of the AKT hydrophobic motif was dramatically can functionally replace the requirement of AKT to support cell elevated, rather than decreased, in mTOR knockout heart tissues, transformation, which suggests that IKKε may act as an AKT indicating the existence of other kinase(s) contributing to AKT regulator or effector. Consistently, IKKε was found to be ampli- phosphorylation. Here we show that the atypical IκB kinase ε and fied in several types of human cancer, including ovarian cancer TANK-binding kinase 1 (IKKε/TBK1) phosphorylate AKT on both the and breast cancer (20, 21). -
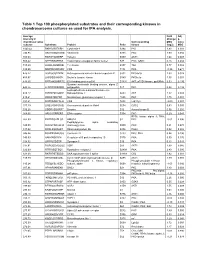
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Members of the Competence Network for Congenital Heart Defects, Germany
Members of the Competence Network for Congenital Heart Defects, Germany Hashim Abdul-Khaliq, Hans-Heiner Kramer, Felix Berger, Brigitte Stiller, Ulrike Bauer, Thomas Pickardt, Sabine Klaassen Family 49 Family 62 Family 226 Family 333 * * * * * * * * TOF AVS ASD ASD PAPVR TOF * * * * ASD PAPVD COA ASD * Family 346 Family 398 VSD * Family 489 * * VSD VSD * * VSD HCM * * * Family 545 BAV AVS ASD PDA Sv AS VSD Family 576 Family 645 Septal defect * * * COA AVS ASD BAV VSD * * * * BAV AVS AVS ASD BAV Family 702 * VSD VSD Family 732 * Family 720 AVSD * * * VSD * * Family 831 TOF TOF TOF Vring TGA VSD * * * * PDA VSD AVS * PDA * * * VSD BAV PFO ASD ASD2 PDA PDA COA Family 1117 Family 1121 * PVS ? * ASD VSD * * * TOF VSD ASD VSD Family 1319 Family 1151 * * * * 2 * ASD ASD * * ASD ASD * * EbA AVS AVS Family 1364 Family 1560 Family 1575 * * TOF VSD ASD VSD infPS TOF * * * VSD PVS PVS PVS VSD * VSD PVS Family 1710 ASD Family 1722 * ASD CHD * VSD VSD * COA TGA DCM ASD BAV SV, MVA COA PVS * DCM DCM ASD HLHS COA BAV Family 2077 Family 2261 * COA, BAV * * * PVS PVS PVS * * infPS infPS BAV ASD * * AVS ASD ASD BAV BAV Family 3500 Family 2558 Family 3315 * * * AVS VSD AVR * * HLHS COA PDA ASD BAV * PVA Family 3501 VSD Family 3503 ? ? * * * VSD HRHS EbA * 3 PVA PAA ASD PDA VSD Family 3505 Family 3540 * * BAV ASD AVS * * * HLHS HLHS TAPVR BAV COA Figure S1. Pedigrees of 32 Danish multiplex CHD families. Circles: females. Squares: males. White symbols: unaffected family members. Filled symbols: affected family members. Triangles: abortion. -

BMK1 Kinase Suppresses Epithelial–Mesenchymal Transition Through the Akt/Gsk3b Signaling Pathway
Published OnlineFirst January 26, 2012; DOI: 10.1158/0008-5472.CAN-11-2055 Cancer Tumor and Stem Cell Biology Research BMK1 Kinase Suppresses Epithelial–Mesenchymal Transition through the Akt/GSK3b Signaling Pathway Runqiang Chen, Qingkai Yang, and Jiing-Dwan Lee Abstract Epithelial–mesenchymal transition (EMT) plays a crucial role in the development of cancer metastasis. The – jun mitogen-activated protein (MAP) kinases extracellular signal regulated kinase, c- -NH2-kinase, and p38 have been implicated in promoting EMT, but a role for the MAP kinase BMK1 has not been studied. Here, we report that BMK1 signaling suppresses EMT. BMK1 elevation augmented E-cadherin–mediated cell–cell adhesion, downregulated mesenchymal markers, and decreased cell motility. Conversely, BMK1 silencing attenuated E-cadherin–mediated cell–cell adhesion, upregulated mesenchymal markers, and stimulated cell motility. BMK1 depletion dramatically increased the accumulation of endogenous Snail in the nuclear compartment. Snail accumulation was mediated by Akt/GSK3b signaling, which was activated by a modulation in the expression of the mTOR inhibitor DEPTOR. In support of these observations, BMK1 depletion promoted metastasis in vivo. Together, our findings reveal a novel mechanism of EMT control via mTOR/Akt inhibition that suppresses cancer metastasis. Cancer Res; 72(6); 1–9. Ó2012 AACR. Introduction that control cancer progression. The majority of mitogenic/ – The process of epithelial–mesenchymal transition (EMT) oncogenic signal activated signaling pathways stimulate is critically involved in the progression of human diseases, EMT(2).Inparticular,3ofthe4mitogen-activatedprotein such as cancer metastasis and fibrosis (1). EMT involves (MAP) kinase pathways described to date (namely, Erk, JNK, profound phenotypic changes that include loss of cell–cell and p38; ref.