Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Reframing Psychiatry for Precision Medicine
Reframing Psychiatry for Precision Medicine Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Recombinant MAP2K2 Protein
Recombinant MAP2K2 protein Catalog No: 81332, 81632 Quantity: 20, 1000 µg Expressed In: Baculovirus Concentration: 0.3 µg/µl Source: Human Buffer Contents: Recombinant MAP2K2 protein is supplied in 25 mM HEPES-NaOH pH 7.5, 300 mM NaCl, 10% glycerol, 0.04% Triton X-100, and 0.5 mM TCEP. Background: MAP2K2 (Mitogen-Activated Protein Kinase Kinase Kinase 2) is a dual specificity protein kinase that belongs to the MAP kinase kinase family. This kinase plays a critical role in mitogen growth factor signal transduction. It phosphorylates and activates MAPK1/ERK2 and MAPK2/ERK3. The activation of this kinase itself is dependent on the Ser/Thr phosphorylation by MAP kinase kinase kinases. Mutations in this gene cause cardiofaciocutaneous syndrome (CFC syndrome), a disease characterized by heart defects, cognitive disability, and distinctive facial features similar to those found in Noonan syndrome. Protein Details: Recombinant MAP2K2 protein was expressed in baculovirus expression system as the full length protein (accession number NP_109587.1) with a N-terminal FLAG Tag. The molecular weight of the protein is 45.7 kDa. Application Notes: This product was manufactured as described in Protein Details. Where possible, Active Motif has developed functional or activity assays for Recombinant MAP2K2 protein gel recombinant proteins. Additional characterization such as enzyme kinetic activity 10% SDS-PAGE with Coomassie blue assays, inhibitor screening or other biological activity assays may not have been staining MW: 45.7 kDa performed for every product. All available data for a given product is shown on the lot- Purity: >90% specific Technical Data Sheet. Storage and Guarantee: Recombinant proteins in solution are temperature sensitive and must be stored at -80°C to prevent degradation. -

Hidden Targets in RAF Signalling Pathways to Block Oncogenic RAS Signalling
G C A T T A C G G C A T genes Review Hidden Targets in RAF Signalling Pathways to Block Oncogenic RAS Signalling Aoife A. Nolan 1, Nourhan K. Aboud 1, Walter Kolch 1,2,* and David Matallanas 1,* 1 Systems Biology Ireland, School of Medicine, University College Dublin, Belfield, Dublin 4, Ireland; [email protected] (A.A.N.); [email protected] (N.K.A.) 2 Conway Institute of Biomolecular & Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland * Correspondence: [email protected] (W.K.); [email protected] (D.M.) Abstract: Oncogenic RAS (Rat sarcoma) mutations drive more than half of human cancers, and RAS inhibition is the holy grail of oncology. Thirty years of relentless efforts and harsh disappointments have taught us about the intricacies of oncogenic RAS signalling that allow us to now get a pharma- cological grip on this elusive protein. The inhibition of effector pathways, such as the RAF-MEK-ERK pathway, has largely proven disappointing. Thus far, most of these efforts were aimed at blocking the activation of ERK. Here, we discuss RAF-dependent pathways that are regulated through RAF functions independent of catalytic activity and their potential role as targets to block oncogenic RAS signalling. We focus on the now well documented roles of RAF kinase-independent functions in apoptosis, cell cycle progression and cell migration. Keywords: RAF kinase-independent; RAS; MST2; ASK; PLK; RHO-α; apoptosis; cell cycle; cancer therapy Citation: Nolan, A.A.; Aboud, N.K.; Kolch, W.; Matallanas, D. Hidden Targets in RAF Signalling Pathways to Block Oncogenic RAS Signalling. -

Establishment and Genomic Characterization of a Sporadic Malignant Peripheral Nerve Sheath Tumor Cell Line Jody Fromm Longo1, Stephanie N
www.nature.com/scientificreports OPEN Establishment and genomic characterization of a sporadic malignant peripheral nerve sheath tumor cell line Jody Fromm Longo1, Stephanie N. Brosius3,5,7, Iya Znoyko1, Victoria A. Alers1, Dorea P. Jenkins1, Robert C. Wilson1,2, Andrew J. Carroll4, Daynna J. Wolf1, Kevin A. Roth6 & Steven L. Carroll1,2,3* Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive Schwann cell-derived neoplasms that occur sporadically or in patients with neurofbromatosis type 1 (NF1). Preclinical research on sporadic MPNSTs has been limited as few cell lines exist. We generated and characterized a new sporadic MPNST cell line, 2XSB, which shares the molecular and genomic features of the parent tumor. These cells have a highly complex karyotype with extensive chromothripsis. 2XSB cells show robust invasive 3-dimensional and clonogenic culture capability and form solid tumors when xenografted into immunodefcient mice. High-density single nucleotide polymorphism array and whole exome sequencing analyses indicate that, unlike NF1-associated MPNSTs, 2XSB cells have intact, functional NF1 alleles with no evidence of mutations in genes encoding components of Polycomb Repressor Complex 2. However, mutations in other genes implicated in MPNST pathogenesis were identifed in 2XSB cells including homozygous deletion of CDKN2A and mutations in TP53 and PTEN. We also identifed mutations in genes not previously associated with MPNSTs but associated with the pathogenesis of other human cancers. These include DNMT1, NUMA1, NTRK1, PDE11A, CSMD3, LRP5 and ACTL9. This sporadic MPNST-derived cell line provides a useful tool for investigating the biology and potential treatment regimens for sporadic MPNSTs. Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive neoplasms derived from the Schwann cell lineage1,2. -

Accumulation of Dephosphorylated 4EBP After Mtor Inhibition with Rapamycin Is Sufficient to Disrupt Paracrine Transformation By
Oncogene (2014) 33, 2405–2412 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc SHORT COMMUNICATION Accumulation of dephosphorylated 4EBP after mTOR inhibition with rapamycin is sufficient to disrupt paracrine transformation by the KSHV vGPCR oncogene D Martin, Q Nguyen, A Molinolo and JS Gutkind Dysregulation of the PI3K/Akt/mTOR pathway is one of the most frequent events in human cancer. However, the clinical benefits of PI3K/Akt/mTOR inhibitors have not yet achieved their predicted potential in many of the most prevalent human cancers. Of interest, treatment of Kaposi’s sarcoma (KS) patients with rapamycin provided the first evidence of the antineoplastic activity of mTOR inhibitors in humans, becoming the standard of care for KS arising in renal transplant patients. Thus, the study of KS may provide a unique opportunity to dissect the contribution of specific mTOR downstream targets to cancer development. The KS- associated herpesvirus (KSHV) is the etiological agent for KS, and the KSHV-encoded oncogene viral-G protein-coupled receptor (vGPCR) promotes the potent activation of the PI3K-Akt-mTOR pathway by both direct and paracrine mechanisms. We focused on a direct target of mTOR, EIF4EBP1/2/3 (4EBP), which inhibits the translation of eukaryotic initiation factor 4E (eiF4E)-bound mRNAs. 4EBP phosphorylation by mTOR relieves its inhibitory activity, hence resulting in increased eiF4E-dependent mRNA translation. We developed a paracrine transformation model, recapitulating the cellular composition of KS lesions, in which vGPCR-expressing cells promote the rapid proliferation of endothelial cells, thus expressing KSHV-latent genes by the release of growth factors. -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -
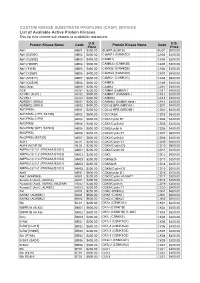
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This List May Change with Respect to Availability and Pricing
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This list may change with respect to availability and pricing. Protein Kinase Name Code U.S. Protein Kinase Name Code U.S. Price Price Abl1 AB01 $200.00 BUBR1(BUB1B) BU01 $600.00 Abl1 [E255K] AB02 $400.00 CaMK1δ (CAMK1D) CA03 $200.00 Abl1 [G250E] AB03 $400.00 CAMK1γ CA04 $200.00 Abl1 [H369P] AB04 $600.00 CAMK2α (CAMK2B) CA05 $200.00 Abl1 [T315I] AB05 $400.00 CaMK2β (CAMK2B) CA06 $200.00 Abl1 [Y253F] AB06 $400.00 CaMK2δ (CAMK2D) CA07 $400.00 Abl1 [M351T] AB07 $600.00 CaMK2γ (CAMK2G) CA08 $600.00 Abl1 [Q252H] AB08 $600.00 CAMK3γ CA09 $200.00 Abl2 (Arg) AB09 $200.00 CAMK4 CA10 $200.00 ACK AC01 $200.00 CAMK4 (CaMKIV ) CA11 $400.00 ACVR1 (ALK2 ) AC02 $400.00 CAMKK1 (CAMKKA ) CA12 $200.00 ACVRL1 AC03 $400.00 CAMKK2 CA13 $200.00 ADRBK1 (GRK2) AD01 $200.00 CAMKK2 (CaMKK beta ) CA14 $400.00 ADRBK2 (GRK3) AD02 $400.00 CDC42 BPA (MRCKA ) CD01 $400.00 Akt1/PKBα AK01 $200.00 CDC42 BPB (MRCKB ) CD02 $400.00 Akt1/PKBα [δPH, S473D] AK02 $600.00 CDC7/ASK CD03 $600.00 Akt1/PKBα [δPH] AK03 $600.00 CDK1/cyclin B1 CD04 $400.00 Akt2/PKBβ AK04 $200.00 CDK1/CyclinA2 CD05 $200.00 Akt2/PKBβ [δPH, S474D] AK05 $600.00 CDK2/cyclin A CD06 $400.00 Akt3/PKBγ AK06 $200.00 CDK2/Cyclin E1 CD07 $600.00 Akt3/PKBγ [S472D] AK07 $600.00 CDK2/CyclinA2 CD08 $200.00 ALK1 AL01 $200.00 CDK3/Cyclin E1 CD09 $600.00 ALK4 (ACVR1B) AL02 $200.00 CDK4//Cyclin D3 CD10 $600.00 AMPKα1/β1/γ1 (PRKAA1/B1/G1) AM01 $200.00 CDK4/Cyclin D1 CD11 $200.00 AMPKα1/β1/γ2 (PRKAA1/B1/G2) AM02 $200.00 CDK5 CD12 $600.00 -

Adaptive Capabilities of the Pi3k/Akt/Mtor Pathway in Acute Myeloid Leukemia Revealed by the Use of Selective Inhibitors
Alma Mater Studiorum – Università di Bologna SCIENZE BIOMEDICHE: PROGETTO 5 “SCIENZE MORFOLOGICHE UMANE E MOLECOLARI” Ciclo XXV SSD : BIO 16, SETTORE CONCORSUALE :05H1 ADAPTIVE CAPABILITIES OF THE PI3K/AKT/MTOR PATHWAY IN ACUTE MYELOID LEUKEMIA REVEALED BY THE USE OF SELECTIVE INHIBITORS Presentata da Jessika Bertacchini Relatori Prof.ssa Lucia Manzoli Prof.ssa Sandra Marmiroli Coordinatore Chiar. mo Prof. Lucio Cocco TABLE OF CONTENTS o ABSTRACT o INTRODUCTION o ACUTE MYELOID LEUKEMIA o PROGNOSIS AND GENETICS o DEREGULATED SIGNAL TRANSDUCTION PATHWAYS IN ACUTE MYELOID LEUKEMIA o PI3K/AKT/MTOR SIGNAL TRANSDUCTION PATHWAY IN ACUTE MYELOID LEUKEMIA o PI3K o AKT/PKB o Mtor o NEGATIVE REGULATION OF PI3K/AKT/mTOR PATHWAY o PI3K/AKT/mTOR PATHWAY AND SURVIVAL o PI3K/AKT/mTOR PATHWAY AND CELL CYCLE o PI3K/AKT/mTOR PATHWAY AND METABOLISM o PI3K/AKT/mTOR INHIBITORS o PI3K INHIBITORS o AKT INHIBITORS o mTOR INHIBITORS o TYROSINE KINASE RECEPTOR o TYROSINE KINASE RECEPTOR IN ACUTE MYELOID LEUKEMIA o AIMS o MATERIALS AND METHODS o PATIENTS DEMOGRAPHICS o CELL CULTURE AND DRUG TREATMENTS o ARRAY ASSEMBLY o RESULTS o RESULTS o DISCUSSION o BIBLIOGRA 1.ABSTRACT The objective of the study was to investigate the sensitivity of primary blasts from AML patients to PI3K/Akt/mTor inhibitors through reverse-phase protein microarray. Reverse-phase microarray assays using phosphospecific antibodies (RPPA) can directly measure levels of phosphorylated protein isoforms. Mapping of deregulated kinases and protein signaling networks within tumors can provide a means to stratify patients with shared biological characteristics to the most optimal treatment, and identify drug targets. In particular, the PI3K/AKT/mTOR signaling pathways are frequently activated in blast cells from patients with acute myelogenous leukemia (AML), a neoplastic disorder characterized by the accumulation of genetically altered myelogenous cells displaying deregulated intracellular signalling pathways and aggressive clinical behavior with poor prognosis. -

Gene Expression Profiling of Micrornas Associated with UCA1 in Bladder Cancer Cells
INTERNATIONAL JOURNAL OF ONCOLOGY 48: 1617-1627, 2016 Gene expression profiling of microRNAs associated with UCA1 in bladder cancer cells XIAOJUAN XIE1,2, JINGJING PAN1, LIQIANG WEI2, SHOUZHEN WU3, HUILIAN HOU4, XU LI3 and WEI CHEN1 1Department of Clinical Laboratory, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710061; 2Shaanxi Center for Clinical Laboratory, Shaanxi Provincial People's Hospital, Xi'an, Shaanxi 710068; 3Center for Translational Medicine, 4Department of Pathology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710061, P.R. China Received November 11, 2015; Accepted December 27, 2015 DOI: 10.3892/ijo.2016.3357 Abstract. Emerging evidence indicates that non-coding and positively correlated with miR-196a, whereas UCA1 and RNAs, such as lncRNAs and microRNAs, play important miR-196a were negatively correlated with p27kip1, which was roles in diverse diseases, such as cancer, immune diseases downregulated in bladder cancer patients. Thus, our findings and cardiovascular diseases. Interestingly, lncRNAs could provided valuable information on miRNAs associated with directly or indirectly regulate the expression of miRNAs. UCA1 in bladder cancer, which could be helpful to further However, the expression profiling of miRNAs associated with explore the related genes and molecular networks fundamental UCA1 in bladder cancer remains unknown. Here, we used in bladder cancer progression. Illumina deep sequencing to sequence miRNA libraries from both the UCA1 knockdown and normal high-expression 5637 Introduction cells. We identified 225 and 235 miRNAs expressed in 5637 cells of normal high-expression and knockdown of UCA1, Recently, emerging studies have demonstrated that 70-90% of respectively. -

Coordinate Phosphorylation of Multiple Residues on Single AKT1 and AKT2 Molecules
Oncogene (2014) 33, 3463–3472 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc ORIGINAL ARTICLE Coordinate phosphorylation of multiple residues on single AKT1 and AKT2 molecules H Guo1,6, M Gao1,6,YLu1, J Liang1, PL Lorenzi2, S Bai3, DH Hawke4,JLi1, T Dogruluk5, KL Scott5, E Jonasch3, GB Mills1 and Z Ding1 Aberrant AKT activation is prevalent across multiple human cancer lineages providing an important new target for therapy. Twenty- two independent phosphorylation sites have been identified on specific AKT isoforms likely contributing to differential isoform regulation. However, the mechanisms regulating phosphorylation of individual AKT isoform molecules have not been elucidated because of the lack of robust approaches able to assess phosphorylation of multiple sites on a single AKT molecule. Using a nanofluidic proteomic immunoassay (NIA), consisting of isoelectric focusing followed by sensitive chemiluminescence detection, we demonstrate that under basal and ligand-induced conditions that the pattern of phosphorylation events is markedly different between AKT1 and AKT2. Indeed, there are at least 12 AKT1 peaks and at least 5 AKT2 peaks consistent with complex combinations of phosphorylation of different sites on individual AKT molecules. Following insulin stimulation, AKT1 was phosphorylated at Thr308 in the T-loop and Ser473 in the hydrophobic domain. In contrast, AKT2 was only phosphorylated at the equivalent sites (Thr309 and Ser474) at low levels. Further, Thr308 and Ser473 phosphorylation occurred predominantly on the same AKT1 molecules, whereas Thr309 and Ser474 were phosphorylated primarily on different AKT2 molecules. Although basal AKT2 phosphorylation was sensitive to inhibition of phosphatidylinositol 3-kinase (PI3K), basal AKT1 phosphorylation was essentially resistant. -

New Insights in RBM20 Cardiomyopathy
Current Heart Failure Reports (2020) 17:234–246 https://doi.org/10.1007/s11897-020-00475-x TRANSLATIONAL RESEARCH IN HEART FAILURE (J BACKS & M VAN DEN HOOGENHOF, SECTION EDITORS) New Insights in RBM20 Cardiomyopathy D. Lennermann1,2 & J. Backs1,2 & M. M. G. van den Hoogenhof1,2 Published online: 13 August 2020 # The Author(s) 2020 Abstract Purpose of Review This review aims to give an update on recent findings related to the cardiac splicing factor RNA-binding motif protein 20 (RBM20) and RBM20 cardiomyopathy, a form of dilated cardiomyopathy caused by mutations in RBM20. Recent Findings While most research on RBM20 splicing targets has focused on titin (TTN), multiple studies over the last years have shown that other splicing targets of RBM20 including Ca2+/calmodulin-dependent kinase IIδ (CAMK2D) might be critically involved in the development of RBM20 cardiomyopathy. In this regard, loss of RBM20 causes an abnormal intracellular calcium handling, which may relate to the arrhythmogenic presentation of RBM20 cardiomyopathy. In addition, RBM20 presents clinically in a highly gender-specific manner, with male patients suffering from an earlier disease onset and a more severe disease progression. Summary Further research on RBM20, and treatment of RBM20 cardiomyopathy, will need to consider both the multitude and relative contribution of the different splicing targets and related pathways, as well as gender differences. Keywords RBM20 . Dilated cardiomyopathy . CaMKIIδ . Calcium handling . Gender differences . Titin Introduction (ARVC), where a small number of genes account for most of the genetic causes, DCM-causing mutations have been ob- Dilated cardiomyopathy (DCM), as defined by left ventricular served in a variety of genes of diverse ontology [2].