TSPY and Its X-Encoded Homologue Interact with Cyclin B but Exert Contrasting Functions on Cyclin-Dependent Kinase 1 Activities
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Reframing Psychiatry for Precision Medicine
Reframing Psychiatry for Precision Medicine Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Characterization of Intellectual Disability and Autism Comorbidity Through Gene Panel Sequencing
bioRxiv preprint doi: https://doi.org/10.1101/545772; this version posted February 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Characterization of Intellectual disability and Autism comorbidity through gene panel sequencing Maria Cristina Aspromonte 1, 2, Mariagrazia Bellini 1, 2, Alessandra Gasparini 3, Marco Carraro 3, Elisa Bettella 1, 2, Roberta Polli 1, 2, Federica Cesca 1, 2, Stefania Bigoni 4, Stefania Boni 5, Ombretta Carlet 6, Susanna Negrin 6, Isabella Mammi 7, Donatella Milani 8 , Angela Peron 9, 10, Stefano Sartori 11, Irene Toldo 11, Fiorenza Soli 12, Licia Turolla 13, Franco Stanzial 14, Francesco Benedicenti 14, Cristina Marino-Buslje 15, Silvio C.E. Tosatto 3, 16, Alessandra Murgia 1, 2, Emanuela Leonardi 1, 2 1. Molecular Genetics of Neurodevelopment, Dept. of Woman and Child Health, University of Padova, Padova, Italy 2. Fondazione Istituto di Ricerca Pediatrica (IRP), Città della Speranza, Padova, Italy 3. Dept. of Biomedical Sciences, University of Padova, Padova, Italy 4. Medical Genetics Unit, Ospedale Universitario S. Anna, Ferrara, Italy 5. Medical Genetics Unit, S. Martino Hospital, Belluno, Italy 6. Child Neuropsychiatry Unit, IRCCS Eugenio Medea, Conegliano, Italy 7. Medical Genetics Unit, Dolo General Hospital, Venezia, Italy 8. Pediatric Highly Intensive Care Unit, Department of Pathophysiology and Transplantation, University of Milano, Fondazione IRCCS, Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy 9. Child Neuropsychiatry Unit, Epilepsy Center, Santi Paolo-Carlo Hospital, Dept. of Health Sciences, University of Milano, Milano, Italy 10. Department of Pediatrics, Division of Medical Genetics, University of Utah School of Medicine, Salt Lake City, UT, USA 11. -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA. -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7, -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5. -
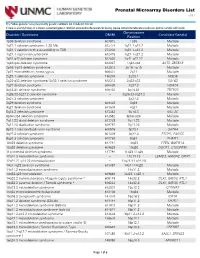
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome -

Two Cyclin-Dependent Kinase Pathways Are Essential for Polarized Trafficking of Presynaptic Components
Two Cyclin-Dependent Kinase Pathways Are Essential for Polarized Trafficking of Presynaptic Components Chan-Yen Ou,1,5 Vivian Y. Poon,2,5 Celine I. Maeder,1 Shigeki Watanabe,3 Emily K. Lehrman,1 Amy K.Y. Fu,4 Mikyoung Park,1 Wing-Yu Fu,4 Erik M. Jorgensen,3 Nancy Y. Ip,4 and Kang Shen1,* 1Department of Biology, Howard Hughes Medical Institute, Stanford University, 385 Serra Mall, California 94305, USA 2Neurosciences Program, Stanford University School of Medicine, 385 Serra Mall, Stanford, California 94305, USA 3Department of Biology, Howard Hughes Medical Institute, University of Utah, Salt Lake City, UT 84112-0840, USA 4Department of Biochemistry, Biotechnology Research Institute and Molecular Neuroscience Center, Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China 5These authors contributed equally to this work *Correspondence: [email protected] DOI 10.1016/j.cell.2010.04.011 SUMMARY Therefore, the localization of synaptic vesicles and other active zone components at axonal presynaptic sites is absolutely Polarized trafficking of synaptic proteins to axons essential for neuronal function. and dendrites is crucial to neuronal function. How are these axonal or dendritic molecules localized appro- Through forward genetic analysis in C. elegans,we priately? There are multiple possible routes that diverge at the identified a cyclin (CYY-1) and a cyclin-dependent levels of sorting, trafficking, and retention (Horton and Ehlers, Pctaire kinase (PCT-1) necessary for targeting 2003). Proteins are sorted into different carrier vesicles in the presynaptic components to the axon. Another cy- cell body. Certain vesicles are specifically transported to one compartment, while others travel into multiple compartments clin-dependent kinase, CDK-5, and its activator before being selectively retained in one compartment. -

Oligonucleotide Microarray Analysis of Distinct Gene Expression Patterns in Colorectal Cancer Tissues Harboring BRAF and K-Ras Mutations
Carcinogenesis vol.27 no.3 pp.392–404, 2006 doi:10.1093/carcin/bgi237 Advance Access publication October 11, 2005 Oligonucleotide microarray analysis of distinct gene expression patterns in colorectal cancer tissues harboring BRAF and K-ras mutations Il-Jin Kim1,y, Hio Chung Kang1,2,y, Sang-Geun Jang1, Introduction Kun Kim1, Sun-A Ahn1, Hyun-Ju Yoon1, Sang Nam Yoon3 and Jae-Gahb Park1,2,3,Ã Colorectal cancer (CRC) is an important human cancer, and incidence rates of this cancer are increasing in Asian countries 1 Korean Hereditary Tumor Registry, Cancer Research Institute and such as Korea (1). CRC development is a multi-step process Downloaded from https://academic.oup.com/carcin/article/27/3/392/2476120 by guest on 27 September 2021 Cancer Research Center, Seoul National University, Seoul, Korea, (2) involving microsatellite instability (MSI), mutations in 2Research Institute and Hospital, National Cancer Center, 809 Madu-dong, Ilsan-gu, Goyang, Gyeonggi 411-764, Korea mismatch repair (MMR) of genes such as MLH1 and MSH2, and 3Department of Surgery, Seoul National University College of and mutations in APC, SMAD4,K-ras, TP53 and b-catenin Medicine, Seoul, Korea (2–9). Somatic BRAF mutations have been reported in ÃTo whom correspondence should be addressed. 5.1–18% of CRCs (10–12). BRAF is one of three serine/ E-mail: [email protected] threonine kinases (ARAF, BRAF, CRAF/RAF1) that act within Various types of human cancers harbor BRAF somatic the RAS–RAF–MEK–ERK–MAPK signaling pathway (13). mutations, leading researchers to seek molecular targets BRAF mutations have also been reported in 80% of primary for BRAF inhibitors. -

Network Impact of the SERT Ala56 Coding Variant
fnmol-13-00089 June 8, 2020 Time: 16:4 # 1 ORIGINAL RESEARCH published: 08 June 2020 doi: 10.3389/fnmol.2020.00089 Ex vivo Quantitative Proteomic Analysis of Serotonin Transporter Interactome: Network Impact of the SERT Ala56 Coding Variant Meagan A. Quinlan1,2,3, Matthew J. Robson4, Ran Ye2, Kristie L. Rose5, Kevin L. Schey5 and Randy D. Blakely2,6* 1 Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA, United States, 2 Department of Pharmacology, Vanderbilt University, Nashville, TN, United States, 3 Department of Biomedical Science, Charles E. Schmidt College of Medicine, Florida Atlantic University, Jupiter, FL, United States, 4 Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, Cincinnati, OH, United States, 5 Department of Biochemistry, Vanderbilt University, Nashville, TN, United States, 6 Brain Institute, Florida Atlantic University, Jupiter, FL, United States Altered serotonin (5-HT) signaling is associated with multiple brain disorders, including major depressive disorder (MDD), obsessive-compulsive disorder (OCD), and autism spectrum disorder (ASD). The presynaptic, high-affinity 5-HT transporter (SERT) tightly regulates 5-HT clearance after release from serotonergic neurons in the brain and enteric nervous systems, among other sites. Accumulating evidence suggests that SERT is dynamically regulated in distinct activity states as a result of environmental and Edited by: Raul R. Gainetdinov, intracellular stimuli, with regulation perturbed by disease-associated coding variants. Saint Petersburg State University, Our lab identified a rare, hypermorphic SERT coding substitution, Gly56Ala, in Russia subjects with ASD, finding that the Ala56 variant stabilizes a high-affinity outward- Reviewed by: facing conformation (SERT∗) that leads to elevated 5-HT uptake in vitro and in vivo. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L.