Epigenetic Loss of the RNA Decapping Enzyme NUDT16 Mediates C-MYC Activation in T-Cell Acute Lymphoblastic Leukemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Integrative Analysis of Phenomic, Genomic, and Transcriptomic To
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.29.392167; this version posted November 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Integrative Analysis of Phenomic, Genomic, and Transcriptomic to Identify Potential Functional Genes of Yaks in Plain and Plateau Jiabo Wang1,2, §, Jiuqiang Guan3, §, Kangzhu Yixi1,2,Tao Shu1, Zhixin Chai1,2, Jikun Wang1,2, Hui Wang1,2, Zhijuan Wu1,2, Xin Cai1,2, Jincheng Zhong1,2*,Xiaolin Luo3* 1. Key Laboratory of Qinghai-Tibetan Plateau Animal Genetic Resource Reservation and Utilization (Southwest Minzu University), Ministry of Education,Chengdu, Sichuan, China; 2. Qinghai-Tibetan Plateau Animal Genetic Resource Reservation and Utilization Key Laboratory of Sichuan Province, Chengdu, Sichuan, China; 3. Sichuan Academy of Grassland Sciences, Chengdu, Sichuan, China; §These authors contributed equally to this work. *Correspondences should be addressed to JZ ([email protected]) and XL ([email protected]) bioRxiv preprint doi: https://doi.org/10.1101/2020.11.29.392167; this version posted November 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Background: The yak is an important source of livelihood for the people living in the Qinghai-Tibet Plateau. Most genetics detection studies have focused on the comparison between different tissues of different breeds, both living in the Plateau and in the plains. The genetic background and complex regulatory relationship have frequently puzzled researchers. In this study, we divided a population of 10 yaks into two subgroups, namely Plateau (living in the Plateau) and Plain (living in the plains). -

Role of DCP1-DCP2 Complex Regulated by Viral and Host Micrornas in DNA Virus Infection T
Fish and Shellfish Immunology 92 (2019) 21–30 Contents lists available at ScienceDirect Fish and Shellfish Immunology journal homepage: www.elsevier.com/locate/fsi Full length article Role of DCP1-DCP2 complex regulated by viral and host microRNAs in DNA virus infection T ∗ Yuechao Sun, Xiaobo Zhang College of Life Sciences and Laboratory for Marine Biology and Biotechnology of Qingdao National Laboratory for Marine Science and Technology, Zhejiang University, Hangzhou, 310058, People's Republic of China ARTICLE INFO ABSTRACT Keywords: The DCP1-DCP2 complex can regulate the antiviral immunity of animals by the decapping of retrovirus RNAs DCP1-DCP2 complex and the suppression of RNAi during RNA virus infection. However, the influence of DCP1-DCP2 complex on DNA miRNA virus infection and the regulation of DCP1-DCP2 complex by microRNAs (miRNAs) remain unclear. In this study, DNA virus infection the role of miRNA-regulated DCP1-DCP2 complex in DNA virus infection was characterized. Our results showed that the DCP1-DCP2 complex played a positive role in the infection of white spot syndrome virus (WSSV), a DNA virus of shrimp. In the DCP1-DCP2 complex, the N-terminal regulatory domain of DCP2 was interacted with the EVH1 domain of DCP1. Furthermore, shrimp miRNA miR-87 inhibited WSSV infection by targeting the host DCP2 gene and viral miRNA WSSV-miR-N46 took a negative effect on WSSV replication by targeting the host DCP1 gene. Therefore, our study provided novel insights into the underlying mechanism of DCP1-DCP2 complex and its regulation by miRNAs in virus-host interactions. Importance: During RNA virus infection, the DCP1-DCP2 complex can play important roles in the animal anti- viral immunity by decapping retrovirus RNAs and suppressing RNAi. -

Atxn2-CAG100-Knockin Mouse Spinal Cord Shows Progressive TDP43
bioRxiv preprint doi: https://doi.org/10.1101/838177; this version posted November 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Atxn2-CAG100-KnockIn mouse spinal cord shows progressive TDP43 pathology associated with cholesterol biosynthesis suppression Júlia Canet-Pons1§, Nesli-Ece Sen1,2§, Aleksandar Arsovic1, Luis-Enrique Almaguer-Mederos1,3, Melanie V. Halbach1, Jana Key1,2, Claudia Döring4, Anja Kerksiek5, Gina Picchiarelli6, Raphaelle Cassel6, Frédérique René6, Stéphane Dieterlé6, Nina Hein-Fuchs7, Renate König7, Luc Dupuis6, Dieter Lütjohann5, Suzana Gispert1, Georg Auburger1# 1 Experimental Neurology, Medical Faculty, Goethe University, 60590 Frankfurt am Main, Germany; 2 Faculty of Biosciences, Goethe University, 60438 Frankfurt am Main, Germany; 3 Center for Investigation and Rehabilitation of Hereditary Ataxias (CIRAH), Holguín, Cuba; 4 Dr. Senckenberg Institute of Pathology, Medical Faculty, Goethe University, 60590 Frankfurt am Main, Germany; 5 Institute for Clinical Chemistry and Clinical Pharmacology, Medical Faculty, University Bonn, 53127 Bonn, Nordrhein-Westfalen, Germany; 6 UMRS-1118 INSERM, Faculty of Medicine, University of Strasbourg, 67000 Strasbourg, France; 7 Host-Pathogen Interactions, Paul-Ehrlich-Institute, 63225 Langen, Germany. § Joint first authorship # Correspondence to: [email protected], Tel: +49-69-6301-7428, FAX: +49-69-6301-7142 Acknowledgements: 1 bioRxiv preprint doi: https://doi.org/10.1101/838177; this version posted November 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Reaction Mechanism of Cellular U Snrnp Assembly
The Reaction Mechanism of Cellular U snRNP Assembly Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Ashwin Chari Aus Bangalore (Indien) Würzburg 2009 Eingereicht am: Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. M. Müller 1. Gutachter: Prof. Dr. U. Fischer 2. Gutachter: Prof. Dr. U. Scheer Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am: Erklärung Erklärung gemäss §4 Absatz 3 der Promotionsordnung der Fakultät für Biologie der Bayerischen Julius-Maximilians-Universität Würzburg vom 15. März 1999 1. Hiermit erkläre ich ehrenwörtlich, dass ich die vorliegende Dissertation selbstständig angefertigt und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe. 2. Ich erkläre, dass die vorliegende Dissertation weder in gleicher noch in ähnlicher Form bereits in einem Prüfungsverfahren vorgelegen hat. 3. Ich erkläre, dass ich ausser den mit dem Zulassungsantrag urkundlich vorgelegten Graden keine weiteren akademischen Grade erworben oder zu erwerben versucht habe. Würzburg, 2009 Ashwin Chari Table of Contents 1. Summary 1 2. Zusammenfassung 5 3. Introduction 9 3.1 Principles Governing Macromolecular Complex Assembly in Vivo 9 3.2 Pre-mRNA Splicing 12 3.3 Architecture of Spliceosomal U snRNPs 14 3.4 The Cell Biology of U snRNP Biogenesis 16 3.5 U snRNP Assembly in Vivo is an Active, Factor-Mediated Process 19 3.6 References 22 4. Goals of this Thesis 29 5. Results 31 5.1 Taking an Inventory of the Subunits of the Human SMN-Complex 31 5.2 Definition of the Basic Architecture of the Human SMN-Complex 49 5.3 Mechanistic Aspects of Cellular U snRNP Assembly 65 5.4 Evolution of the SMN-Complex 115 6. -
C a B D G E F
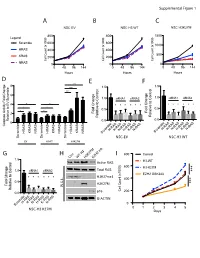
Supplemental Figure 1 A B C NSC EV NSC H3 WT NSC H3K27M 800 800 1500 ) ) Legend ) Scramble 600 600 x1000 x1000 x1000 ( ( ( 1000 t t t n n HRAS n 400 400 u u u o o o C C C 500 l l KRAS l 200 200 el el el C C C NRAS 0 0 0 0 48 96 144 0 48 96 144 0 48 96 144 Hours Hours Hours D *** E F 8 1.5 1.5 e *** l l *** ro ro t 6 t 1.0 siRNA1 siRNA2 1.0 siRNA1 siRNA2 Con Con o o t 4 * * t * * * * * * e * * * * * * e v i * * v i t 0.5 * * t 0.5 Fold Change 2 Fold Change Rela Rela Relative to EV Scrambl 0.0 0.0 0 Caspase Activity Fold Change H-RASK-RASN-RASH-RASK-RASN-RAS H-RASK-RASN-RASH-RASK-RASN-RAS KRAS KRAS KRAS HRAS NRAS HRAS NRAS HRAS NRAS Scramble Scramble Scramble Scramble Scramble NSC-EV NSC-H3 WT EV H3WT H3K27M G H I 800 Control Con WT H3 H3K27MEZH2 inb 1.5 l H3-WT Active RAS ro ) t 600 H3-K27M 1.0 siRNA1 siRNA2 Total RAS *** Con x1000 ( o EZH2 GSK343 t t * * * * * * H3K27me3 n e 400 u v i o t 0.5 *** H3K27M C Fold Change l W.C.L. el Rela p16 C 200 0.0 B-ACTIN H-RASK-RASN-RASH-RASK-RASN-RAS 0 Scramble NSC-H3 K27M 0 1 2 3 4 5 Days E C A 100 Cell Viability 100 25 50 75 Cell Viability 25 50 75 0 0 Fold Change MOCK MOCK Relative to Control Scramble * 0.0 0.2 0.4 0.6 0.8 1.0 1.2 MYC Scramble MYC MYC * PDGFRA Scramble siRNA PDGFRA MYC PDGFRA AURKA AURKA DIPG007siRNA2 PDGFRA NSC H3K27MsiRNA1and AURKA LAMTOR3 AURKA LAMTOR3 LAMTOR3 LIN28A LAMTOR3 LIN28A LIN28B LIN28A NSC-EV LIN28A MAP2K3 LIN28B LIN28B LIN28B MAP2K5 siRNA1 MAP2K3 MAP2K3 MAP3K2 MAP2K3 MAP3K7 MAP2K5 MAP2K5 MAPK7 MAP2K5 2 MAP3K2 siRNA1 siRNA2 MAP3K2 ZAK MAP3K7 MAP3K2 MAP3K7 MAPK7 MAP3K7 -

Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in Vitro and in Vivo
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Genetik Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in vitro and in vivo - Towards Response Prediction in Cancer Therapy with Kinase Inhibitors Michaela Bairlein Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ. -Prof. Dr. K. Schneitz Prüfer der Dissertation: 1. Univ.-Prof. Dr. A. Gierl 2. Hon.-Prof. Dr. h.c. A. Ullrich (Eberhard-Karls-Universität Tübingen) 3. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 07.01.2010 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 19.04.2010 angenommen. FOR MY PARENTS 1 Contents 2 Summary ................................................................................................................................................................... 5 3 Zusammenfassung .................................................................................................................................................... 6 4 Introduction .............................................................................................................................................................. 8 4.1 Cancer .............................................................................................................................................................. -

A Non-Canonical Role for the EDC4 Decapping Factor in Regulating
RESEARCH ARTICLE A non-canonical role for the EDC4 decapping factor in regulating MARF1- mediated mRNA decay William R Brothers1, Steven Hebert1, Claudia L Kleinman1,2, Marc R Fabian1,3,4* 1Lady Davis Institute for Medical Research, Jewish General Hospital, Montreal, Canada; 2Department of Human Genetics, McGill University, Montreal, Canada; 3Department of Biochemistry, McGill University, Montreal, Canada; 4Department of Oncology, McGill University, Montreal, Canada Abstract EDC4 is a core component of processing (P)-bodies that binds the DCP2 decapping enzyme and stimulates mRNA decay. EDC4 also interacts with mammalian MARF1, a recently identified endoribonuclease that promotes oogenesis and contains a number of RNA binding domains, including two RRMs and multiple LOTUS domains. How EDC4 regulates MARF1 action and the identity of MARF1 target mRNAs is not known. Our transcriptome-wide analysis identifies bona fide MARF1 target mRNAs and indicates that MARF1 predominantly binds their 3’ UTRs via its LOTUS domains to promote their decay. We also show that a MARF1 RRM plays an essential role in enhancing its endonuclease activity. Importantly, we establish that EDC4 impairs MARF1 activity by preventing its LOTUS domains from binding target mRNAs. Thus, EDC4 not only serves as an enhancer of mRNA turnover that binds DCP2, but also as a repressor that binds MARF1 to prevent the decay of MARF1 target mRNAs. Introduction *For correspondence: The regulated decay of eukaryotic mRNA populations plays an important role in the post-transcrip- [email protected] tional control (PTC) of gene expression. These PTC programs are, in turn, critical for regulating a number of biological processes, including during early development, cell proliferation and immune Competing interests: The response. -

Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S
crossmark Research Author’s Choice © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S Rolf Turk‡§¶ʈ**, Jordy J. Hsiao¶, Melinda M. Smits¶, Brandon H. Ng¶, Tyler C. Pospisil‡§¶ʈ**, Kayla S. Jones‡§¶ʈ**, Kevin P. Campbell‡§¶ʈ**, and Michael E. Wright¶‡‡ Mutations in genes encoding components of the sar- The muscular dystrophies are hereditary diseases charac- colemmal dystrophin-glycoprotein complex (DGC) are re- terized primarily by the progressive degeneration and weak- sponsible for a large number of muscular dystrophies. As ness of skeletal muscle. Most are caused by deficiencies in such, molecular dissection of the DGC is expected to both proteins associated with the cell membrane (i.e. the sarco- reveal pathological mechanisms, and provides a biologi- lemma in skeletal muscle), and typical features include insta- cal framework for validating new DGC components. Es- bility of the sarcolemma and consequent death of the myofi- tablishment of the molecular composition of plasma- ber (1). membrane protein complexes has been hampered by a One class of muscular dystrophies is caused by mutations lack of suitable biochemical approaches. Here we present in genes that encode components of the sarcolemmal dys- an analytical workflow based upon the principles of pro- tein correlation profiling that has enabled us to model the trophin-glycoprotein complex (DGC). In differentiated skeletal molecular composition of the DGC in mouse skeletal mus- muscle, this structure links the extracellular matrix to the cle. We also report our analysis of protein complexes in intracellular cytoskeleton. -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior.