An Exploration of Mutation Status of Cancer Genes in Breast Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

1 Tumor Suppressor PLK2 May Serve As a Biomarker in Triple-Negative Breast Cancer for Improved Response to PLK1 Therapeutics
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.448722; this version posted June 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Tumor suppressor PLK2 may serve as a biomarker in triple-negative breast cancer for improved response to PLK1 therapeutics Yang Gao1, 2, 3, Elena B. Kabotyanski1, 2, Elizabeth Villegas7, Jonathan H. Shepherd8, Deanna Acosta1, 2, Clark Hamor1, 2, Tingting Sun2,4,5, Celina Montmeyor-Garcia9, Xiaping He8, Lacey E. Dobrolecki1, 2, 3, Thomas F. Westbrook2, 4, 5, Michael T. Lewis1, 2, 3, Susan G. Hilsenbeck2, 3, Xiang H.-F. Zhang1, 2, 3, 6, Charles M. Perou8 and Jeffrey M. Rosen1, 2 1Department of Molecular and Cellular Biology 2Dan L. Duncan Cancer Center 3Lester and Sue Smith Breast Center 4Department of Molecular and Human Genetics 5Verna & Marrs McLean Department of Biochemistry and Molecular Biology 6McNair Medical Institute Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA 7University of Houston-Downtown, Houston, TX 77002, USA 8The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA 9 Canadian Blood Services, Toronto, ON M5G 2M1, Canada Correspondence to Jeffrey M. Rosen (Mail Stop: BCM130, Room: BCM-M638a, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030. Office: 713-798-6210. Fax: 713-898-8012. Email: [email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.448722; this version posted June 16, 2021. -

Genome-Wide Association Study to Identify Genomic Regions And
www.nature.com/scientificreports OPEN Genome‑wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle H. Sweett1, P. A. S. Fonseca1, A. Suárez‑Vega1, A. Livernois1,2, F. Miglior1 & A. Cánovas1* Fertility plays a key role in the success of calf production, but there is evidence that reproductive efciency in beef cattle has decreased during the past half‑century worldwide. Therefore, identifying animals with superior fertility could signifcantly impact cow‑calf production efciency. The objective of this research was to identify candidate regions afecting bull fertility in beef cattle and positional candidate genes annotated within these regions. A GWAS using a weighted single‑step genomic BLUP approach was performed on 265 crossbred beef bulls to identify markers associated with scrotal circumference (SC) and sperm motility (SM). Eight windows containing 32 positional candidate genes and fve windows containing 28 positional candidate genes explained more than 1% of the genetic variance for SC and SM, respectively. These windows were selected to perform gene annotation, QTL enrichment, and functional analyses. Functional candidate gene prioritization analysis revealed 14 prioritized candidate genes for SC of which MAP3K1 and VIP were previously found to play roles in male fertility. A diferent set of 14 prioritized genes were identifed for SM and fve were previously identifed as regulators of male fertility (SOD2, TCP1, PACRG, SPEF2, PRLR). Signifcant enrichment results were identifed for fertility and body conformation QTLs within the candidate windows. Gene ontology enrichment analysis including biological processes, molecular functions, and cellular components revealed signifcant GO terms associated with male fertility. -

Kinase Profiling Book
Custom and Pre-Selected Kinase Prof iling to f it your Budget and Needs! As of July 1, 2021 19.8653 mm 128 196 12 Tyrosine Serine/Threonine Lipid Kinases Kinases Kinases Carna Biosciences, Inc. 2007 Carna Biosciences, Inc. Profiling Assays available from Carna Biosciences, Inc. As of July 1, 2021 Page Kinase Name Assay Platform Page Kinase Name Assay Platform 4 ABL(ABL1) MSA 21 EGFR[T790M/C797S/L858R] MSA 4 ABL(ABL1)[E255K] MSA 21 EGFR[T790M/L858R] MSA 4 ABL(ABL1)[T315I] MSA 21 EPHA1 MSA 4 ACK(TNK2) MSA 21 EPHA2 MSA 4 AKT1 MSA 21 EPHA3 MSA 5 AKT2 MSA 22 EPHA4 MSA 5 AKT3 MSA 22 EPHA5 MSA 5 ALK MSA 22 EPHA6 MSA 5 ALK[C1156Y] MSA 22 EPHA7 MSA 5 ALK[F1174L] MSA 22 EPHA8 MSA 6 ALK[G1202R] MSA 23 EPHB1 MSA 6 ALK[G1269A] MSA 23 EPHB2 MSA 6 ALK[L1196M] MSA 23 EPHB3 MSA 6 ALK[R1275Q] MSA 23 EPHB4 MSA 6 ALK[T1151_L1152insT] MSA 23 Erk1(MAPK3) MSA 7 EML4-ALK MSA 24 Erk2(MAPK1) MSA 7 NPM1-ALK MSA 24 Erk5(MAPK7) MSA 7 AMPKα1/β1/γ1(PRKAA1/B1/G1) MSA 24 FAK(PTK2) MSA 7 AMPKα2/β1/γ1(PRKAA2/B1/G1) MSA 24 FER MSA 7 ARG(ABL2) MSA 24 FES MSA 8 AurA(AURKA) MSA 25 FGFR1 MSA 8 AurA(AURKA)/TPX2 MSA 25 FGFR1[V561M] MSA 8 AurB(AURKB)/INCENP MSA 25 FGFR2 MSA 8 AurC(AURKC) MSA 25 FGFR2[V564I] MSA 8 AXL MSA 25 FGFR3 MSA 9 BLK MSA 26 FGFR3[K650E] MSA 9 BMX MSA 26 FGFR3[K650M] MSA 9 BRK(PTK6) MSA 26 FGFR3[V555L] MSA 9 BRSK1 MSA 26 FGFR3[V555M] MSA 9 BRSK2 MSA 26 FGFR4 MSA 10 BTK MSA 27 FGFR4[N535K] MSA 10 BTK[C481S] MSA 27 FGFR4[V550E] MSA 10 BUB1/BUB3 MSA 27 FGFR4[V550L] MSA 10 CaMK1α(CAMK1) MSA 27 FGR MSA 10 CaMK1δ(CAMK1D) MSA 27 FLT1 MSA 11 CaMK2α(CAMK2A) MSA 28 -

Modulation of NF-Κb Signalling by Microbial Pathogens
REVIEWS Modulation of NF‑κB signalling by microbial pathogens Masmudur M. Rahman and Grant McFadden Abstract | The nuclear factor-κB (NF‑κB) family of transcription factors plays a central part in the host response to infection by microbial pathogens, by orchestrating the innate and acquired host immune responses. The NF‑κB proteins are activated by diverse signalling pathways that originate from many different cellular receptors and sensors. Many successful pathogens have acquired sophisticated mechanisms to regulate the NF‑κB signalling pathways by deploying subversive proteins or hijacking the host signalling molecules. Here, we describe the mechanisms by which viruses and bacteria micromanage the host NF‑κB signalling circuitry to favour the continued survival of the pathogen. The nuclear factor-κB (NF-κB) family of transcription Signalling targets upstream of NF‑κB factors regulates the expression of hundreds of genes that NF-κB proteins are tightly regulated in both the cyto- are associated with diverse cellular processes, such as pro- plasm and the nucleus6. Under normal physiological liferation, differentiation and death, as well as innate and conditions, NF‑κB complexes remain inactive in the adaptive immune responses. The mammalian NF‑κB cytoplasm through a direct interaction with proteins proteins are members of the Rel domain-containing pro- of the inhibitor of NF-κB (IκB) family, including IκBα, tein family: RELA (also known as p65), RELB, c‑REL, IκBβ and IκBε (also known as NF-κBIα, NF-κBIβ and the NF-κB p105 subunit (also known as NF‑κB1; which NF-κBIε, respectively); IκB proteins mask the nuclear is cleaved into the p50 subunit) and the NF-κB p100 localization domains in the NF‑κB complex, thus subunit (also known as NF‑κB2; which is cleaved into retaining the transcription complex in the cytoplasm. -

Comparing Signaling Networks Between Normal and Transformed Hepatocytes Using Discrete Logical Models
Published OnlineFirst July 8, 2011; DOI: 10.1158/0008-5472.CAN-10-4453 Cancer Integrated Systems and Technologies: Mathematical Oncology Research Comparing Signaling Networks between Normal and Transformed Hepatocytes Using Discrete Logical Models Julio Saez-Rodriguez1,2, Leonidas G. Alexopoulos1,2, MingSheng Zhang1, Melody K. Morris2, Douglas A. Lauffenburger2, and Peter K. Sorger1,2 Abstract Substantial effort in recent years has been devoted to constructing and analyzing large-scale gene and protein networks on the basis of "omic" data and literature mining. These interaction graphs provide valuable insight into the topologies of complex biological networks but are rarely context specific and cannot be used to predict the responses of cell signaling proteins to specific ligands or drugs. Conversely, traditional approaches to analyzing cell signaling are narrow in scope and cannot easily make use of network-level data. Here, we combine network analysis and functional experimentation by using a hybrid approach in which graphs are converted into simple mathematical models that can be trained against biochemical data. Specifically, we created Boolean logic models of immediate-early signaling in liver cells by training a literature-based prior knowledge network against biochemical data obtained from primary human hepatocytes and 4 hepatocellular carcinoma cell lines exposed to combinations of cytokines and small-molecule kinase inhibitors. Distinct families of models were recovered for each cell type, and these families clustered topologically into normal and diseased sets. Cancer Res; 71(16); 5400–11. Ó2011 AACR. Introduction Major Findings The availability of high-throughput interaction data has led Clustering arises from systematic differences in signaling to the creation of methods for summarizing and exploring logic in three regions of the network. -

MAP2K4/MKK4 Expression in Pancreatic Cancer: Genetic Validation of Immunohistochemistry and Relationship to Disease Course
8516 Vol. 10, 8516–8520, December 15, 2004 Clinical Cancer Research MAP2K4/MKK4 Expression in Pancreatic Cancer: Genetic Validation of Immunohistochemistry and Relationship to Disease Course Wei Xin,1 Ki J. Yun,4 Francesca Ricci,2 ing patterns were also evaluated among unresectable pri- Marianna Zahurak,3 Wanglong Qiu,4 mary and metastatic cancer tissues from autopsy specimens, Gloria H. Su,4 Charles J. Yeo,5,6 indicating intact Mkk4 immunolabeling in 88.8% of the unresectable primary carcinomas as compared with 63.3% Ralph H. Hruban,4,5 Scott E. Kern,4,5 and 4,5 of distant metastases (P < 0.001). Our data indicate that the Christine A. Iacobuzio-Donahue loss of Mkk4 protein expression in pancreatic carcinomas 1 Department of Pathology, The University of Michigan Medical may be more frequent than suggested by the rates of genetic Center, Ann Arbor, Michigan; 2Department of Pathology, University La Sapienza, Rome, Italy; and the Departments of 3Biostatistics, inactivation alone and that MKK4 loss may contribute to 4Pathology, 5Oncology, and 6Surgery, The Johns Hopkins University disease progression. The correlation of MKK4 genetic status Hospital, Baltimore, Maryland with immunolabeling patterns validate this approach for the evaluation of MKK4 status in routine histologic sections and ABSTRACT may provide useful information regarding patient prognosis. MKK4 (MAP2K4/SEK1) is a member of the mitogen- activated protein kinase family, originally identified as a INTRODUCTION kinase involved in the stress-activated protein kinase path- The mitogen-activated protein (MAP) kinase cascades are way by directly phosphorylating c-Jun NH -terminal kinase. 2 multifunctional signaling pathways that are evolutionally well MKK4 genetic inactivation has been observed in a subset of conserved in all of the eukaryotic cells. -
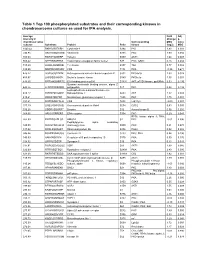
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Protein-Protein Interactions Among Signaling Pathways May Become New Therapeutic Targets in Liver Cancer (Review)
ONCOLOGY REPORTS 35: 625-638, 2016 Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer (Review) XIAO ZHANG1*, YULAN WANG1*, Jiayi WANG1,2 and FENYONG SUN1 1Department of Clinical Laboratory Medicine, Shanghai Tenth People's Hospital of Tongji University, Shanghai 200072; 2Translation Medicine of High Institute, Tongji University, Shanghai 200092, P.R. China Received May 29, 2015; Accepted July 6, 2015 DOI: 10.3892/or.2015.4464 Abstract. Numerous signaling pathways have been shown to be 1. Introduction dysregulated in liver cancer. In addition, some protein-protein interactions are prerequisite for the uncontrolled activation Liver cancer is the sixth most common cancer and the second or inhibition of these signaling pathways. For instance, in most common cause of cancer-associated mortality world- the PI3K/AKT signaling pathway, protein AKT binds with wide (1). Approximately 75% of all primary liver cancer types a number of proteins such as mTOR, FOXO1 and MDM2 to are hepatocellular carcinoma (HCC) that formed from liver play an oncogenic role in liver cancer. The aim of the present cells. Liver cancer can be formed from other structures in review was to focus on a series of important protein-protein the liver such as bile duct, blood vessels and immune cells. interactions that can serve as potential therapeutic targets Secondary liver cancer is a result of metastasis of cancer from in liver cancer among certain important pro-carcinogenic other body sites into the liver. The major cause of primary liver signaling pathways. The strategies of how to investigate and cancer is viral infection with either hepatitis C virus (HCV) analyze the protein-protein interactions are also included in or hepatitis B virus (HBV), which leads to massive inflamma- this review. -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5. -

The Landscape of MAP3K1/MAP2K4 Alterations in Gastrointestinal (GI) Malignancies
The landscape of MAP3K1/MAP2K4 alterations in gastrointestinal (GI) malignancies. Matthew K. Stein1,2, Andrew Elliott3, Jimmy J. Hwang4, Emil Lou5, Moh’d M. Khushman6, Aaron James Scott7, John L. Marshall8, Davendra Sohal9, Benjamin A. Weinberg8, Richard M. Goldberg10, Mohamed E. Salem4, W. Michael Korn3,11, Axel Grothey2 1. West Cancer Center, Germantown, TN; 2. University of Tennessee Health Science Center, Memphis, TN; 3. CARIS Life Sciences, Irving, TX; 4. Levine Cancer Institute, Charlotte, NC; 5. University of Minnesota School of Medicine, Minneapolis, MN; 6. Medical Oncology, The University of South Alabama, Mitchell Cancer Institute, Mobile, AL; 7. Banner-University of Arizona Cancer Center, Division of Hematology and Oncology, Tucson, AZ; 8. Georgetown Lombardi Comprehensive Cancer Center, Washington, DC; 9. University of Cincinnati, Cincinnati, OH; 10. West Virginia University Cancer Institute, Morgantown, WV; 11. University of California San Francisco, San Francisco, CA; 2. West Cancer Center, Germantown, TN Results Table 2. – Frequency of MAP3K1/MAP2K4-MT by GI malignancy site. Results, continued Background Table 1. – Patient characteristics MAP3K1 % MAP3K1 and/or and/or Figure 4. – Comparison of selected co-mutation rates of MAP3K1/MAP2K4-MT versus WT • Inactivating alterations in MAP3K1/MAP2K4 occur in various solid CRC CRC Non-CRC Non-CRC Total MAP2K4- MAP2K4- non-CRC patients (all MSS except as noted). All GI MAP3K1/ MAP3K1/ MAP3K1/ MAP3K1/ Characteristic Cancer Type cases mutated mutated 70.0% tumors, sensitize cancer -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain