Loudon Chapter 11 Review: Ethers & Epoxides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -
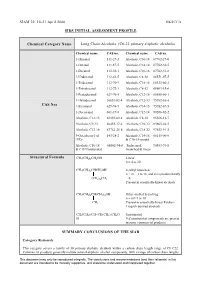
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Transcription 12.01.12
Lecture 2B • 01/12/12 We covered three different reactions for converting alcohols into leaving groups. One was to turn an alcohol into an alkyl chloride, that was using thionyl chloride. Second reaction was using tosyl chloride; the primary difference between those two reactions is one of stereochemistry. An inversion of stereochemistry does occur if you use thionyl chloride, because it does affect the carbon-oxygen bond, but because forming a tosylate does not touch the carbon-oxygen bond, only the oxygen- hydrogen bond, there’s no change in stereochemistry there. We then saw phosphorus tribromide that reacts very similarly to the thionyl chloride; you get an alkyl bromide instead, but it also has inversion of configuration. The last reaction is not a new mechanism, it is just an Sn2 reaction, it’s called the Finkelstein reaction. Really this works, in a sense, off of Le Châtelier’s principle. In solution, in theory, sodium iodide can displace bromide, but sodium bromide can displace iodide, so you can have an Sn2 reaction that goes back and forth and back and forth and back and forth. Except, sodium iodide is somewhat soluble in acetone, while sodium chloride and sodium bromide are not. So, in fact, one of the things that we get out of this as a by-product is sodium bromide, which, again, is not soluble in acetone and therefore precipitates out and is no longer part of the reaction mixture, so there’s no reverse reaction possible. Because of this solubility trick, it allows this reaction to be pulled forward, which means you can get the alkyl iodide. -

Reactions of Alcohols & Ethers 1
REACTIONS OF ALCOHOLS & ETHERS 1. Combustion (Extreme Oxidation) alcohol + oxygen carbon dioxide + water 2 CH3CH2OH + 6 O2 4 CO2 + 6 H2O 2. Elimination (Dehydration) ° alcohol H 2 S O 4/ 1 0 0 C alkene + water H2SO4/100 ° C CH3CH2CH2OH CH3CH=CH2 + H2O 3. Condensation ° excess alcohol H 2 S O 4 / 1 4 0 C ether + water H2SO4/140 ° C 2 CH3CH2OH CH3CH2OCH2CH3 + H2O 4. Substitution Lucas Reagent alcohol + hydrogen halide Z n C l2 alkyl halide + water ZnCl2 CH3CH2OH + HCl CH3CH2Cl + H2O • This reaction with the Lucas Reagent (ZnCl2) is a qualitative test for the different types of alcohols because the rate of the reaction differs greatly for a primary, secondary and tertiary alcohol. • The difference in rates is due to the solubility of the resulting alkyl halides • Tertiary Alcohol→ turns cloudy immediately (the alkyl halide is not soluble in water and precipitates out) • Secondary Alcohol → turns cloudy after 5 minutes • Primary Alcohol → takes much longer than 5 minutes to turn cloudy 5. Oxidation • Uses an oxidizing agent such as potassium permanganate (KMnO4) or potassium dichromate (K2Cr2O7). • This reaction can also be used as a qualitative test for the different types of alcohols because there is a distinct colour change. dichromate → chromium 3+ (orange) → (green) permanganate → manganese (IV) oxide (purple) → (brown) Tertiary Alcohol not oxidized under normal conditions CH3 KMnO4 H3C C OH NO REACTION K2Cr2O7 CH3 tertbutyl alcohol Secondary Alcohol ketone + hydrogen ions H O KMnO4 H3C C CH3 + K2Cr2O7 C + 2 H H3C CH3 OH propanone 2-propanol Primary Alcohol aldehyde + water carboxylic acid + hydrogen ions O KMnO4 O H H KMnO H H 4 + CH3CH2CH2OH C C C H C C C H + 2 H K2Cr2O7 + H2O K Cr O H H H 2 2 7 HO H H 1-propanol propanal propanoic acid 6. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Soln-2212-PT3-S20-Ch-16-18.Pdf
1. 2. H2SO4 H2SO4 H2SO41) BH3, THF 1) BH3, THF 1) BH3, THF - - - 2) OH , H2O2, H2O2) OH , H2O2, H2O 2) OH , H2O2, H2O OH OH OH 3. OH OH OH H2SO4 1) BH3, THF - 2) OH , H2O2, H2O OH PBr3 PBr3 PBr3 Pyridine Pyridine Pyridine OH PBr3 Pyridine O O O Mg Mg Mg HO HO HO1) 1) 1) Ether Ether Ether 2) HCl 2) HCl 2) HCl Br Br Br MgBr MgBr MgBr O Mg HO 1) Ether 2) HCl Br 4. MgBr **This reaction reduces an aldehyde to a primary alcohol and a ketone to a secondary alcohol. Since only one EQ of reagent is given the most reactive functional group will be reduced (in this case the aldehyde) **ketones are less reactive than aldehydes and the more steric hindrance surrounding the ketone the less reactive it is. Note: NaBH4 reduces ketones but is too weak to reduce an ester/double/triple bonds 5. ** With these reagents a tertiary amide will be reduced to a tertiary amine, a secondary amide will be reduced to a secondary amine, and a primary amide will be reduced to a primary amine 6. ** Recall from chapter 7, OChem 1 H2 and Lindlars catalyst always reduces an alkyne to a CIS ALKENE ** Li/Na and liquid NH3 always reduces an alkyne to a TRANS ALKENE 7. **A ketone/aldehyde reacts with primary amines in trace acid to produce an imine, and reacts with a secondary amine in trace acid to produce an enamine (double bond forms between the alpha and beta carbons) **Sodium cyanoborohydride (NaBH3CN) is used to reduce imines/enamines back to their respective secondary/tertiary amines 8. -

Chapter 19 the Chemistry of Aldehydes and Ketones. Addition Reactions
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 19 The Chemistry of Aldehydes and Ketones. Addition Reactions Solutions to In-Text Problems 19.1 (b) (d) (e) (g) 19.2 (a) 2-Propanone (d) (E)-3-Ethoxy-2-propenal (f) 4,4-Dimethyl-2,5-cyclohexadienone 19.3 (b) 2-Cyclohexenone has a lower carbonyl stretching frequency because its two double bonds are conjugated. 19.4 (b) The compound is 2-butanone: (c) The high frequency of the carbonyl absorption suggests a strained ring. (See Eq. 19.4, text p. 897.) In fact, cyclobutanone matches the IR stretching frequency perfectly and the NMR fits as well: 19.6 The structure and CMR assignments of 2-ethylbutanal are shown below. The two methyl groups are chemically equivalent, and the two methylene groups are chemically equivalent; all carbons with different CMR chemical shifts are chemically nonequivalent. INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 19 2 19.7 (a) The double bonds in 2-cyclohexenone are conjugated, but the double bonds in 3-cyclohexenone are not. Consequently, 2-cyclohexenone has the UV spectrum with the greater lmax. 19.9 Compound A, vanillin, should have a p T p* absorption at a greater lmax when dissolved in NaOH solution because the resulting phenolate can delocalize into the carboxaldehyde group; the resulting phenolate from compound B, isovanillin, on the other hand, can only delocalize in the aromatic ring. 19.11 The mass spectrum of 2-heptanone should have major peaks at m/z = 43 (from a-cleavage), 71 (from inductive cleavage), and 58 (from McLafferty rearrangement). -

Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and Tert-Amyl Alcohol: a Density Functional Study
Equilibrium Structures and Vibrational Assignments for Isoamyl Alcohol and tert-Amyl Alcohol: A Density Functional Study Wolfgang Forner¨ and Hassan M. Badawi Department of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia Reprint requests to Wolfgang Forner.¨ E-mail: [email protected] Z. Naturforsch. 2013, 68b, 841 – 851 / DOI: 10.5560/ZNB.2013-3003 Received February 9, 2013 We have calculated the vibrational spectra of isoamyl alcohol and of tert-amyl alcohol using Den- sity Functional Theory (DFT) with the Becke-3 Lee Yang Parr (B3LYP) functional and a 6-311+G** atomic basis set. The energies of the conformers were also calculated with ab initio Perturbation Theory of second (MP2) and fourth order restricted to single, double and quadruple excitations (MP4 SDQ) with the same basis set. We found rather complicated equilibria of four conformations in each case, counting only those with appreciable abundancies. PED data were compared with GAUSSVIEW animations. The calculated wavenumbers agree rather well with the experimental ones when the gauche-trans conformer is assumed as the most important one for isoamyl alcohol, and the gauche- gauche one for tert-amyl alcohol. However, some of the experimental bands had to be assigned also to other conformers, indicating their presence in the equilibrium mixture. Due to sterical reasons both the CO and the OH bonds appear to be weaker in the tertiary alcohol, considering the wavenumbers of the CO and OH bond stretching vibrations. The bond lengths point into the same direction, however, the OH bond in the tertiary alcohol is only slightly longer than that in the primary alcohol. -

United States Patent (19) 11 Patent Number: 5,380,763 Sato, Deceased Et Al
USO05380763A United States Patent (19) 11 Patent Number: 5,380,763 Sato, deceased et al. 45) Date of Patent: Jan. 10, 1995 54 TOPICAL COMPOSITION FORTREATING ACNEWU LGARIS FOREIGN PATENT DOCUMENTS 013459 7/1980 European Pat. Off........ A6K 7/48 75) Inventors: Toshiya Sato, deceased, late of 0331489 9/1989 European Pat. Off........ A6K 7/48 Kanagawa, by Kuniko Sato, legal 60-1524-15 8/1985 Japan ......................... A61K 3/045 representative; Kenya Ishida, 63-27425 2/1988 Japan ......................... A61K 3/045 Kanagawa, all of Japan 1551637 8/1979 United Kingdom .......... A61K.7/48 OTHER PUBLICATIONS 73 Assignee: Takasago International Corporation, Adachietal I CA. 105: 11857x (1986) of JP 6105014 10 Tokyo, Japan Jan. 1986. Adachietal. II CA. 104:155727g (1986) of JP 61 15869 (21) Appl. No.: 154,458 23 Jan. 1986. Adachietal. III CA 1.02:137579g (1985) of EP 1297782 22 Filed: Nov. 19, 1993 Jan. 1985. Oono et al. CA 106:201540g (1987) of Ger DE 3522853 30 Foreign Application Priority Data 2 Jan. 1986. Nov. 19, 1992 JP Japan .................................. 4-332232 N. Kato et al., J. Antibact. Antifung. Agents, 8(8), 1-7 (1980). 51) Int. Cl............................................. A61K 31/045 M. Hattoriet al., Chem. Pharm. Bull, 35(8), 3507-3510 52 U.S. Cl. .............. ... 514/724; 514/844; (1987). 514/859 Primary Examiner-Shep K. Rose 58) Field of Search ........................ 514/724, 844,859 Attorney, Agent, or Firn-Sughrue, Mion, Zinn, Macpeak & Seas 56 References Cited (57) ABSTRACT U.S. PATENT DOCUMENTS A topical composition for treating acne vulgaris com 3,806,593 4/1974 Swanbeck et al. -

Organic Synthesis
PART B ORGANIC SYNTHESIS 2:14 PM 1 What is Synthesis? Synthesis involves combining two or more chemical entities through covalent bonds to generate a more complex molecule. The covalent bonds formed between these chemical entities could be based on: a substitution reaction, an addition reaction Since the new bonds formed during synthesis occur at functional groups inherent in polyfunctional molecules, it is usually necessary to use protecting groups to minimize the 2:14 PM formation of side-products. 2 Is Synthesis of Any Value? Synthesis is important to the food and cosmetics industry, but also finds wide application in medicine. Although nature can provide the source of cinnamaldehyde (occurs in the cinnamon plant), its wide application as a flavouring agent requires that it be readily available in large quantities. Synthesis has served to supplement the natural source. 2:14 PM 3 Of What Value is Synthesis? Synthesis of Moisturizers and Sweeteners Enzymatic hydrolysis of starch provides glucose, which when reduced provides sorbitol, a moisturizer and sweetner. 2:14 PM 4 Organic Synthesis What are the Essentials in Synthesis? Since organic synthesis is applied organic chemistry, to stand a realistic chance of succeeding in any synthesis, the student ought to have a good knowledge-base of organic chemistry in the following areas: Protecting group chemistry Asymmetric synthesis Functional group transformations (SCH 202 & SCH 206) Substitution reactions (SCH 102) Addition reactions (SCH 202 and SCH 206) 2:14 PM Strategies for synthetic planning (Retrosynthetic analysis) 5 Protecting Groups in Organic Synthesis 6 2:14 PM Challenges in Organic Synthesis Chemoselectivity can be Elusive Selective functionalization of poly-functional molecules is an important attribute in multi-step organic synthesis. -

Reactions of Alcohols
Reactions of Alcohols Alcohols are versatile organic compounds since they undergo a wide variety of transformations – the majority of which are either oxidation or reduction type reactions. Normally: Oxidation is a loss of electrons; Reduction is a gain of electrons. But in organic terms: Oxidation: loss of H2; addition of O or O2; addition of X2 (halogens). Reduction: - addition of H2 or H ; loss of O or O2; loss of X2. Neither an oxidation nor reduction: Addition or loss of H+, H2O, HX. Ch11 Reacns of Alcohols (landscape).docx Page 1 Oxidation of Alcohols Primary and secondary alcohols are easily oxidized by a variety of reagents. Secondary Alcohols The most common reagent used for oxidation of secondary alcohols to ketones is chromic acid, H2CrO4. Chromic acid is produced in situ by reaction of sodium dichromate, sulfuric acid and water. Na2Cr2O7 + H2O + 2H2SO4 2 H2CrO4 + 2 NaHSO4 Ch11 Reacns of Alcohols (landscape).docx Page 2 Mechanism of oxidation The alcohol and chromic acid produce a chromate ester, which then reductively eliminates the Cr species. The Cr is reduced (VI IV), the alcohol is oxidized. Oxidation of Primary Alcohols Primary alcohols are easily oxidized just like secondary alcohols, and the INITIAL product of oxidation is an aldehyde. Ch11 Reacns of Alcohols (landscape).docx Page 3 However, the aldehyde can also be easily oxidized to an acid, and this ‘over-oxidation’ is a practical problem. E.g. A common reagent that selectively oxidizes a primary alcohol to an aldehyde (and no further) is pyridinium chlorochromate, PCC. N: CrO3, HCl (PCC) E.g. Tertiary Alcohols These are resistant to oxidation because they have no hydrogen atoms attached to the oxygen bearing carbon (carbinol carbon). -

Chapter 10: Structure and Synthesis of Alcohols
Chapter 10: Structure and Synthesis of Alcohols 100 Physical Properties Alcohols can be: OH CH3 CH3 CH CH2CH3 * CH3 CH CH2OH * Secondary alcohol Primary alcohol CH3 * CH3 C OH CH3 Tertiary alcohol 101 • Most alcohols (C 1-C12 ) are liquids at room temperature • Bp much higher than alkanes of similar molecular weights (H-bonding) 102 Acidity : the H of the hydroxyl group is weakly acidic: 103 This hydroxyl proton can be removed with suitable bases. Bases such as KOH or NaOH are not strong enough. NaH, KH or Na (s) , K (s) are usually used. 104 In the case of phenols, the pKa of the hydroxyl group is around 10 and weaker bases can be used (NaOH, KOH, etc…) 105 Synthesis of Alcohols (Review) § Hydration (a) Direct (8.4) 106 Mechanism of Direct Hydration of Alkenes 107 (b) Indirect (4.8) (1) Oxymercuration/Demercuration (8.5) (2) Hydroboration/Oxidation (8.7) 108 § Hydroxylation (8.14) 109 New Synthesis of Alcohols (Nu attack on carbonyl compounds) An excellent method to make alcohols. Depending on the type of carbonyl used, primary, secondary and tertiary alcohols are all available from this reaction. 110 2 types of reagents can deliver nucleophilic carbons to C=O: ◦ Grignard reagent: R-Mg-X ◦ Alkyl Lithium: R-Li δ− δ+ δ− δ+ R M gX R Li 111 § Synthesis of Grignard Reagents (10.8) Br ether MgBr + Mg Cl MgCl ether CH3CHCH2CH3 + Mg CH3CHCH2CH3 112 Alkyl Lithium can be prepared in a similar way: 113 § Synthesis of 1 o Alcohols (10.9) Grignards or alkyl lithium react with formaldehyde to produce primary alcohols.