Development of an Assay for Determining PDE5 Enzyme Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shale Gas and Groundwater Quality
Shale Gas and Groundwater Quality A literature review on fate and effects of added chemicals Alette Langenhoff 1202141-008 © Deltares, 2011 1202141-008-ZWS-0001, 28 December 2011, final Contents 1 Introduction 1 2 The process of fracturing or fracking 5 3 The use of chemicals 7 4 Polyacrylamide 8 4.1 Aerobic degradation of polyacrylamide 8 4.2 Anaerobic degradation 10 4.3 Chemical or physical removal 10 4.4 Conclusion on removal of polyacrylamide 10 5 Glutaraldehyde 12 5.1 Biocide 12 5.2 Biodegradation 12 5.3 Chemical inactivation of glutaraldehyde 13 6 Conclusions 14 7 References 15 Appendices 17 Appendices A Chemicals identified in hydraulic fracturing fluid and flowback/produced water (EPA, 2011). A-1 B Fracturing fluid ingredients and common uses (Europe Unconventional Gas 2011) B-1 C Properties of Polyacrylamide (source: Wikipedia) C-1 D Properties of Glutaraldehyde (source: Wikipedia) D-1 Shale Gas and Groundwater Quality i 1202141-008-ZWS-0001, 28 December 2011, final 1 Introduction Shale gas is a so-called unconventional sources of natural gas, and is one of the most rapidly expanding trends in onshore domestic oil and gas exploration and production today (Fig. 1 and 2). Shale gas is present in hydrocarbon rich shale formations. Shallow gas is commonly defined as gas occurrences in unconsolidated sediments of Tertiary age (often down to depths of 1000 m below surface). The occurrences are positively associated with thick Neogene sediments and are often trapped in anticlinal structures associated with rising salt domes (Muntendam-Bos et al, 2009). Shale has low matrix permeability, so gas production in commercial quantities requires fractures to provide permeability. -
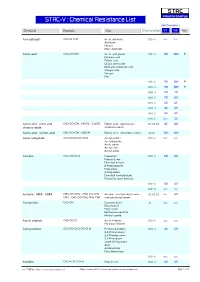
STAC-V : Chemical Resistance List Max Temperature
S TA C Industrial Coatings STAC-V : Chemical Resistance List Max Temperature Chemical Formula Alias Concentration V1 V2 Note Acetaldehyde CH3-CH=O Acetic aldehyde 100 % n.r. n.r. Aldehyde Ethanal Ethyl aldehyde Acetic acid CH3-CO-OH Acetic acid glacial 010 % 90 100 0 Ethanoic acid Ethylic acid Glacial acetic acid Methane carboxylic acid Vinegar acid Vinegar Hac 015 % 90 100 0 025 % 90 100 0 040 % 80 90 050 % 70 80 075 % 60 65 080 % 45 45 085 % 45 45 100 % n.r. 25 Acetic acid : nitric acid : CH3-CO-OH : HNO3 : Cr2O3 Ethylic acid : salpeterzuur : 03:05:03 65 80 chromic oxide chromium oxide Acetic acid : sulfuric acid CH3-CO-OH : H2SO4 Ethylic acid : dihydrogen sulfate 20:10 100 100 Acetic anhydride CH3-CO-O-CO-CH3 Acetyl acetate 100 % n.r. n.r. Acetanhydride Acetic oxide Acetyl ether Acetyl oxide Acetone CH3-CO-CH3 Propanone 005 % 80 80 Propan-2-one Dimethyl ketone β-Ketopropane[ Propanone 2-Propanone Dimethyl formaldehyde Pyroacetic spirit (archaic) 010 % 80 80 100 % n.r. n.r. Acetone : MEK : MiBK CH3-CO-CH3 : CH3-CO-CH2- Acetone : methylethyl ketone : 02:02:02 n.r. 40 CH3 : CH3-CO-CH2-CH2-CH3 methylisobutyl ketone Acetonitrile CH3-CN Cyanomethane all n.r. n.r. Ethanenitrile Ethyl nitrile Methanecarbonitrile Methyl cyanid Acetyl chloride CH3-CO-Cl Acetic chloride 100 % n.r. n.r. Ethanoyl chloride Acetylacetone CH3-CO-CH2-CO-CH3 Pentane-2,4-dione 020 % 40 50 2,4-Pentanedione 2,4-Dioxopentane 2,4-Pentadione acetyl-2-Propanone Acac Acetoacetone Diacetylmethane 100 % n.r. -

Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC
Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC R E S E A R C H A N D D E V E L O P M E N T EPA/600/R-06/105 September 2006 Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC By S. H. Hilal Ecosystems Research Division National Exposure Research Laboratory Athens, Georgia U.S. Environmental Protection Agency Office of Research and Development Washington, DC 20460 NOTICE The information in this document has been funded by the United States Environmental Protection Agency. It has been subjected to the Agency's peer and administrative review, and has been approved for publication. Mention of trade names of commercial products does not constitute endorsement or recommendation for use. ii ABSTRACT SPARC (SPARC Performs Automated Reasoning in Chemistry) chemical reactivity models were extended to calculate hydrolysis rate constants for carboxylic acid ester and phosphate ester compounds in aqueous non- aqueous and systems strictly from molecular structure. The energy differences between the initial state and the transition state for a molecule of interest are factored into internal and external mechanistic perturbation components. The internal perturbations quantify the interactions of the appended perturber (P) with the reaction center (C). These internal perturbations are factored into SPARC’s mechanistic components of electrostatic and resonance effects. External perturbations quantify the solute-solvent interactions (solvation energy) and are factored into H-bonding, field stabilization and steric effects. These models have been tested using 1471 reliable measured base, acid and general base-catalyzed carboxylic acid ester hydrolysis rate constants in water and in mixed solvent systems at different temperatures. -

I Norgan Ic C He Mi Str Y
View Article Online / Journal Homepage / Table of Contents for this issue INORGANIC CHEMISTRY. 443 I n o r g a n ic C h e mi s t r y. Composition of Atmospheres which Extinguish Flame. By FRANKCLOWES ( PTOC. Roy. Soc., 1894, 56, 2-6) .--The experimental flame burning at a platinum jet 1 mm. in diameter, was 0.75 in. in height; it was gradually lowered into a cylinder containing the atmosphere of mixed gases, and these were considered to be in extinctive proportions if the flame was extinguished during its downward passage, or immediately on attaining its lowest position in the cylinder. The gaseous mixture was regarded as containing the minimum quantitr of extinctive gas, when the flame on being lowered into another mixture containing 1 per cent. less of such gas continued to burn in it for a few seconds before being ex- tinguished. Experiments made with flames of hydrogen and Published on 01 January 1895. Downloaded 25/10/2014 07:12:09. alcohol, varying from 0.4 in. to 1.5 in. in height, show that the varying dimensions of the flame are without influence on the proportion of carbonic anhydride in the air necessary to pro- duce extinction. Characteristic differences were observed between the behaviour of wick-fed flames and that of gas-fed flames when they were introduced into an atmosphere which extinguished them, the wick-fed flames gradually diminishing in size until they vanished, whilst the gas-fed flames gradually increased in size, becoming paler and apparently lower in temperature until they suddenly expired. -

The Iron-Dependent Cyanide and Hydrogen Peroxide Co-Toxicity in Escherichia Coli and Its Catastrophic Consequences for the Chromosome
THE IRON-DEPENDENT CYANIDE AND HYDROGEN PEROXIDE CO-TOXICITY IN ESCHERICHIA COLI AND ITS CATASTROPHIC CONSEQUENCES FOR THE CHROMOSOME BY TULIP MAHASETH DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Microbiology in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Andrei Kuzminov, Chair and Director of Research Professor John E. Cronan Professor Jeffrey F. Gardner Associate Professor Carin K. Vanderpool ABSTRACT 2+ Hydrogen peroxide (H2O2) can oxidize cytoplasmic ferrous ions (Fe ) to produce highly reactive hydroxyl radicals (•OH) via Fenton’s reaction that can damage various biomolecules causing oxidative stress. Even though at concentrations higher than 20 mM H2O2 by itself can efficiently kill micro-organisms, it is metabolically impossible for eukaryotic cells to generate H2O2, an uncharged molecule, in such large quantities inside the cell. We propose that potentiation of physiologically relevant amounts of H2O2 by various small molecules serves as a more feasible and safe mechanism to combat invading microbes. NO potentiation of H2O2 toxicity is a known bactericidal weapon employed by macrophages. In fact, in human neutrophils activated by bacterial infection, the myeloperoxidase enzyme catalyzes the formation of hydrogen cyanide (HCN) from serum thiocyanate (SCN-). In the past, researchers have reported that a combination of low millimolar doses of H2O2 and cyanide (CN), which are individually bacteriostatic, caused rapid synergistic killing in Escherichia coli. Our aim is to understand the immune cells antimicrobial responses by investigating the mechanism of CN potentiation of H2O2 toxicity and its chromosomal consequences. We have found that the ability of CN to recruit iron from intracellular depots such as ferritin contributes to its potentiation of H2O2 toxicity, whereas the major stationary phase intracellular iron depot protein, Dps, can sequester this iron, thereby quelling Fenton's reaction. -

Bioorganic Chemistry
Hermann Dugas Christopher Penney Bioorganic Chemistry A Chemical Approach to Enzyme Action With 82 Figures Spri nger-Verlag New York Heidelberg Berlin Dr. Hermann Dugas Dr. Christopher Penney Departement de Chimie Connaught Research Institute Universite de Montreal Willowdale, Ontario Montreal, Quebec Canada M2N 5T8 Canada H3C 3Vl Series Editor: Prof. Charles R. Cantor Columbia University Box 608 Havemeyer Hall New York, New York 10027 USA Cover: The green illustration represents the hypothetical mode of binding of a rigid structural analogue of N-benzoyl-L-phenylalanine methyl ester at the active site of a-chymotrypsin. The illustration emphasizes the equilibration toward the favored configuration (see text page 224). The background design is taken from a diagrammatic representation of the primary structure of a-chymotrypsin. After Nature with permission [B.W. Matthews, P.B. Sigler, R. Henderson, and D.M. Blow (1967), Nature 214, 652-656]. Library of Congress Cataloging in Publication Data Dugas, Hermann, 1942- Bioorganic chemistry. (Springer advanced texts in chemistry.) Bibliography: p. Includes index. 1. Enzymes. 2. Biological chemistry. 3. Chemistry,. Organic. I. Penney, Christopher, 1950- joint author. II. Title. m. Series. [DNLM: 1. Biochemistry. 2. Enzymes-Metabolism. QUl35 D866b] QP60 1. D78 574.19'25 80-16222 All rights reserved. No part of this book may be translated or reproduced in any form without written permission from Springer-Verlag. The use of general descriptive names, trade names, trademarks, etc. in this publication, even if the former are not especially identified, is not to be taken as a sign that such names, as understood by the Trade Marks and Merchandise Marks Act, may accordingly be used freely by anyone. -

Introduction to Fertilizers Industries
Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org ADANA UNIVERSTY – INDUSTRY JOINT RESEARCH CENTER INTRODUCTIN TO FERTILIZER INDUSTRIES BY TAREK ISMAIL KAKHIA 1 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org ADANA UNIVERSTY – INDUSTRY JOINT RESEARCH CENTER page Item 3 Fertilizer 71 N - P - K rating 17 Fertilizers Inorganic Acids: 21 Sulfur 53 Sulfur Dioxide 11 Sulfur Trioxide 44 Sulfuric Acid 35 Nitrogen 35 Liquid Nitrogen 37 Nitrogen Cycle 51 Nitrogen Oxide 53 Nitric oxide ( NOX ) 22 Nitrogen Di Oxide 27 Nitrous Oxide 111 Nitric Oxide 121 Di Nitrogen Pent oxide 714 Nitric Acid 151 Phosphate Minerals ( Phosphate Rock (Phospharite ٌ 157 155 Phosphorus 132 Phosphorus Oxide 171 Phosphorus Tri Oxide 172 Phosphorus Pent Oxide 711 Phosphoric Acid 137 Phospho Gypsum 711 Fertilizers Alkalizes: 787 Ammonia 211 Ammonia Production 211 Amine Gas Treating 171 Ammonium Hydroxide 212 Category : Ammonium Compounds 221 Potassium 253 Potassium Hydroxide 211 Fertilizers Salts 215 Ammonium Ferric Citrate 2 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org ADANA UNIVERSTY – INDUSTRY JOINT RESEARCH CENTER 211 Ammonium Nitrate 215 Di Ammonium Phosphate 212 Tri Ammonium Phosphate 231 Ammonium Sulfate 232 Calcium Nitrate 231 Calcium Phosphate 233 Mono Calcium Phosphate 271 Di Calcium Phosphate 271 Tri Calcium Phosphate 271 Sodium Nitrate 267 Magnesium Phosphate 275 Di Magnesium Phosphate 272 Magnesium Sulfate 235 Potassium Chloride 235 Potassium Citrate 251 Potassium Nitrate 253 Potassium Phosphate 257 Mono Potassium Phosphate 255 Di Potassium Phosphate 252 Tri Potassium Phosphate 221 Potassium Sulfate 221 Borax 511 Organic Fertilizers: 515 Compost 513 Composting 521 Urea 555 Urea Cycle 555 Urea Phosphate 331 Extension & Supplements 511 Macronutrient & Micronutrient Fertilizers 535 Category : Phosphate minerals 533 Pozzolan 535 Pumice 5 Copyright © Tarek Kakhia. -

(Difluoromethylene)Triphosphoric Acid
Synthesis and biological evaluation of fluorinated deoxynucleotide analogs based on bis- (difluoromethylene)triphosphoric acid G. K. Surya Prakasha,1, Mikhail Zibinskya, Thomas G. Uptona, Boris A. Kashemirova, Charles E. McKennaa, Keriann Oertella, Myron F. Goodmana, Vinod K. Batrab, Lars C. Pedersenb, William A. Beardb, David D. Shockb, Samuel H. Wilsonb, and George A. Olaha,1 aLoker Hydrocarbon Research Institute, Department of Chemistry and Department of Biology, University of Southern California, 837 Bloom Walk, Los Angeles, CA 90089-1661; and bLaboratory of Structural Biology, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709 Contributed by George A. Olah, June 3, 2010 (sent for review April 28, 2010) It is difficult to overestimate the importance of nucleoside tri- the altered chemical properties conferred on the compound by phosphates in cellular chemistry: They are the building blocks the fluorine substituent. The van der Waals’ radius of the fluorine for DNA and RNA and important sources of energy. Modifications atom (1.47 Å) is close to the size of hydrogen (1.2 Å). Most of the of biologically important organic molecules with fluorine are of other substituent groups often used to replace hydrogen in the great interest to chemists and biologists because the size and creation of analogs are much larger. Thus fluorine is of electronegativity of the fluorine atom can be used to make defined unique value in the design of analogs, which can very closely ap- structural -

LIGAND FORMULA INDEX Order of Elements: C,Il,O,N, Others In
LIGAND FORMULA INDEX Order of elements: C,Il,O,N, others in alphabetical order. - AsF6 74 HO 1 Br - 115 HOBr 134 Br6Ir 135 HOC1 134 HOI 134 CHON 28 H02N 47 CHN 26 H0 2C1 134 CHNS 29 H031 126 CH203 37 H04NF 2S2 135 - CH2N2 135 H0 4S 79 CH2S3 131 H0 4Se 93 CH2S4 131 H0 4Tc 135 CH2Se3 131 HF 96 CNSe 35 HN3 45 C2N3 36 H20Z 75 C4H204Fe 135 H2OZN2 53, 135 - C4N3 36 H203N2 135 C5H05Mn 135 H203FP 132 C6H4 N6Fe 21 H203S 78 3- C6N6Co 24 HZ03SZ 86 3- C6N6Fe 22 HZ03Se 91 C8H3N8W 135 H204Cr 17 C8H4N8W 135 H204Mn 135 H204Mo 18 C1 104 H204S2 135 3- C1 6Ir 135 H204W 19 H205S 133 F6P 74 H208S2 89 253 254 LIGAND FORMULA INDEX H2S 76 H4N2 43 H2S4 133 H50NlS 132 H2S5 133 H502Nl 132 H2Se 90 H505Sb 133 H2Te 94 H505Ta 135 H30N 44 H506NP 2 71 H302P 54 H5061 129 H303NS 88 H501oP3 63 H303As 132 H606N3P3 72 H303B 25 H606Te 134 H30l 55 H6013P4 66 H30lS 132 H6018P6 70 H304NS 133 H708Nl3 71 H304As 133 H7016P5 135 H304P 56 H8019Nb 6 131 H304V 15 H8019P6 135 H305P 131 H8024P8 70 H309P3 68 H1604l14 135 H3N 40 H620121P60 135 HlS4 132 H403NP 132 I 122 H404Ge 131 H404Si 39 ON 135 H 0 Te - 4 4 95 °3N 48 H 0 P 4 6 2 72, 73 °3Br 121 H 0 P S 4 6 2 2 135 °3C1 113 H 0 P 2- 4 7 2 59 °3SSe 88 H 0 P 4 8 2 73 °4C1 114 H 0 FP - 4 9 3 135 °4Mn 135 H 0 P 69 - 4 12 4 °4Re 20 LIGAND NAME INDEX Ammonia, 40 Hydrogen amidophosphate, 132 Antimonic acid, 133 Hydrogen amidosu1fate, 88 Arsenic acid, 133 Hydrogen antimonate, 133 Arsenous acid, 132 Hydrogen arsenate, 133 Hydrogen arsenite, 132 Boric acid, 25 Hydrogen azide, 45 Bromate ion, 121 Hydrogen borate, 25 Bromide ion, 115 Hydrogen carbonate, -

The Stability and Nature of Complexes of Mercury with Higher Phosphates
This dissertation has been 63-2552 microfilmed exactly as received SIMONAITIS, Richard Ambrose, 1930- THE STABILITY AND NATURE OF COMPLEXES OF MERCURY WITH HIGHER PHOSPHATES. The Ohio State University, Ph.D., 1962 Chemistry, analytical University Microfilms, Inc., Ann Arbor, Michigan THE STABILITY AND HATHRE OF COMPLEXES OF MERCURY WITH HIGHER PHOSPHATES DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Richard Ambrose Simonaitis, B.S., M.Sc. The Ohio State University 1962 Approved by A dviser ipartment of Chemistry ACKNOWLEDGMENTS The author wishes to express his great appreciation to his adviser. Professor James I. Watters, for h lB great interest, under standing and guidance in all phases of this work. He also wishes to thank his wife, Vera, for her aid in the preparation of this manuscript. i i CONTENTS CHAPTER Page I INTRODUCTION .............................................................................. 1 II METHOD..................................................................................... 21 III THE COMPLEXES OF MERCURY(I) WITH PYROPHOSPHATE . 31 Experimental .......................................................... 31 Experimental Results and Discussion .................... 33 IV THE COMPLEXES OF MERCURY(I) WITH TRIPHOSPHATE . 6l Experimental .......................................................... 6 l The Acidity Constants of Triphosphoric Acid in the Presence of 1 M Sodium and Potassium Ions .................................................................................... -

(CDR) by CASRN Or Accession Number
List of Chemicals Reported for the 2012 Chemical Data Reporting (CDR) by CASRN or Accession Number For the 2012 CDR, 7,674 unique chemicals were reported by manufacturers (including importers). Chemicals are listed by CAS Registry Number (for non-confidential chemicals) or by TSCA Accession Number (for chemicals listed on the confidential portion of the TSCA Inventory). CASRN or CASRN or ACCESSION ACCESSION NUMBER CA INDEX NAME or GENERIC NAME NUMBER CA INDEX NAME or GENERIC NAME 100016 Benzenamine, 4-nitro- 10042769 Nitric acid, strontium salt (2:1) 10006287 Silicic acid (H2SiO3), potassium salt (1:2) 10043013 Sulfuric acid, aluminum salt (3:2) 1000824 Urea, N-(hydroxymethyl)- 10043115 Boron nitride (BN) 100107 Benzaldehyde, 4-(dimethylamino)- 10043353 Boric acid (H3BO3) 1001354728 4-Octanol, 3-amino- 10043524 Calcium chloride (CaCl2) 100174 Benzene, 1-methoxy-4-nitro- 100436 Pyridine, 4-ethenyl- 10017568 Ethanol, 2,2',2''-nitrilotris-, phosphate (1:?) 10043842 Phosphinic acid, manganese(2+) salt (2:1) 2,7-Anthracenedisulfonic acid, 9,10-dihydro- 100447 Benzene, (chloromethyl)- 10017591 9,10-dioxo-, sodium salt (1:?) 10045951 Nitric acid, neodymium(3+) salt (3:1) 100185 Benzene, 1,4-bis(1-methylethyl)- 100469 Benzenemethanamine 100209 1,4-Benzenedicarbonyl dichloride 100470 Benzonitrile 100210 1,4-Benzenedicarboxylic acid 100481 4-Pyridinecarbonitrile 10022318 Nitric acid, barium salt (2:1) 10048983 Phosphoric acid, barium salt (1:1) 9-Octadecenoic acid (9Z)-, 2-methylpropyl 10049044 Chlorine oxide (ClO2) 10024472 ester Phosphoric acid, -

United States Patent Office Patented Jan
2,700,038 United States Patent Office Patented Jan. 18, 1955 2 tating with alcohol, the salts which separate would cor respond to the pH-composition titration curves. To our 2,700,038 Suprise, this has not been found to be the case, the di basic salts being precipitated from strongly acidified solu DIBASIC SALTS OF ADENOSINE TRIPHOSPHATE tions. This may be illustrated as follows: On adding AND METHOD OF PREPARATION either sodium, potassium or lithium chloride to an acidi Samuel H. Lipton, Milwaukee, and Samuel A. Morell, fied aqueous solution of the free adenosine triphosphoric Whitefish Bay, Wis., assignors to Pabst Brewing Com acid which is free of cations other than hydrogen ion, and pany, Milwaukee, Wis., a corporation of Delaware then adding an alcohol such as ethanol, methanol or pro O panol, the diabasic salts separate and are found to be No Drawing. Application September 27, 1951, free of chloride ion. For example, a 5% solution of Serial No. 248,632 adenosine triphosphoric acid was adjusted to pH 1.0 by adding hydrochloric acids, then sodium chloride was add 13 Claims. (C. 260-211.5) ed to 1% concentration and the solution was poured into 4 volumes of ethanol. After filtering, washing with adenosineThis invention triphosphoric relates acid, to amore new particularlyand useful asalt water of ethanol and drying, the product was found to be disodium soluble dibasic salt, and to a method for the preparation dihydrogen adenosine triphosphate of high purity and thereof. free of chloride ion. As is well known in the art (P. Ostern, U.