(Difluoromethylene)Triphosphoric Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sulfonyl-Containing Nucleoside Phosphotriesters And
Sulfonyl-Containing Nucleoside Phosphotriesters and Phosphoramidates as Novel Anticancer Prodrugs of 5-Fluoro-2´-Deoxyuridine-5´- Monophosphate (FdUMP) Yuan-Wan Sun, Kun-Ming Chen, and Chul-Hoon Kwon†,* †Department of Pharmaceutical Sciences, College of Pharmacy and Allied Health Professions, St. John’s University, Jamaica, New York 11439 * To whom correspondence should be addressed. Department of Pharmaceutical Sciences, College of Pharmacy and Allied Health Professions, St. John’s University, 8000 Utopia parkway, Jamaica, NY 11439. Tel: (718)-990-5214, fax: (718)-990-6551, e-mail: [email protected]. Abstract A series of sulfonyl-containing 5-fluoro-2´-deoxyuridine (FdU) phosphotriester and phosphoramidate analogues were designed and synthesized as anticancer prodrugs of FdUMP. Stability studies have demonstrated that these compounds underwent pH dependent β-elimination to liberate the corresponding nucleotide species with half-lives in the range of 0.33 to 12.23 h under model physiological conditions in 0.1M phosphate buffer at pH 7.4 and 37 °C. Acceleration of the elimination was observed in the presence of human plasma. Compounds with FdUMP moiety (4-9) were considerably more potent than those without (1-3) as well as 5-fluorouracil (5-FU) against Chinese hamster lung fibroblasts (V-79 cells) in vitro. Addition of thymidine (10 µM) reversed the growth inhibition activities of only 5-FU and the compounds with FdUMP moiety, but had no effect on those without. These results suggested a mechanism of action of the prodrugs involving the intracellular release of FdUMP. Introduction 5-Fluoro-2´-deoxyuridine-5´-monophosphate (FdUMP) is the major metabolite responsible for the anticancer activity of 5-FU (Chart 1). -

(12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub
US 20050044778A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0044778A1 Orr (43) Pub. Date: Mar. 3, 2005 (54) FUEL COMPOSITIONS EMPLOYING Publication Classification CATALYST COMBUSTION STRUCTURE (51) Int. CI.' ........ C10L 1/28; C1OL 1/24; C1OL 1/18; (76) Inventor: William C. Orr, Denver, CO (US) C1OL 1/12; C1OL 1/26 Correspondence Address: (52) U.S. Cl. ................. 44/320; 44/435; 44/378; 44/388; HOGAN & HARTSON LLP 44/385; 44/444; 44/443 ONE TABOR CENTER, SUITE 1500 1200 SEVENTEENTH ST DENVER, CO 80202 (US) (57) ABSTRACT (21) Appl. No.: 10/722,127 Metallic vapor phase fuel compositions relating to a broad (22) Filed: Nov. 24, 2003 Spectrum of pollution reducing, improved combustion per Related U.S. Application Data formance, and enhanced Stability fuel compositions for use in jet, aviation, turbine, diesel, gasoline, and other combus (63) Continuation-in-part of application No. 08/986,891, tion applications include co-combustion agents preferably filed on Dec. 8, 1997, now Pat. No. 6,652,608. including trimethoxymethylsilane. Patent Application Publication Mar. 3, 2005 US 2005/0044778A1 FIGURE 1 CALCULATING BUNSEN BURNER LAMINAR FLAME VELOCITY (LFV) OR BURNING VELOCITY (BV) CONVENTIONAL FLAME LUMINOUS FLAME Method For Calculating Bunsen Burner Laminar Flame Velocity (LHV) or Burning Velocity Requires Inside Laminar Cone Angle (0) and The Gas Velocity (Vg). LFV = A, SIN 2 x VG US 2005/0044778A1 Mar. 3, 2005 FUEL COMPOSITIONS EMPLOYING CATALYST Chart of Elements (CAS version), and mixture, wherein said COMBUSTION STRUCTURE element or derivative compound, is combustible, and option 0001) The present invention is a CIP of my U.S. -

Shale Gas and Groundwater Quality
Shale Gas and Groundwater Quality A literature review on fate and effects of added chemicals Alette Langenhoff 1202141-008 © Deltares, 2011 1202141-008-ZWS-0001, 28 December 2011, final Contents 1 Introduction 1 2 The process of fracturing or fracking 5 3 The use of chemicals 7 4 Polyacrylamide 8 4.1 Aerobic degradation of polyacrylamide 8 4.2 Anaerobic degradation 10 4.3 Chemical or physical removal 10 4.4 Conclusion on removal of polyacrylamide 10 5 Glutaraldehyde 12 5.1 Biocide 12 5.2 Biodegradation 12 5.3 Chemical inactivation of glutaraldehyde 13 6 Conclusions 14 7 References 15 Appendices 17 Appendices A Chemicals identified in hydraulic fracturing fluid and flowback/produced water (EPA, 2011). A-1 B Fracturing fluid ingredients and common uses (Europe Unconventional Gas 2011) B-1 C Properties of Polyacrylamide (source: Wikipedia) C-1 D Properties of Glutaraldehyde (source: Wikipedia) D-1 Shale Gas and Groundwater Quality i 1202141-008-ZWS-0001, 28 December 2011, final 1 Introduction Shale gas is a so-called unconventional sources of natural gas, and is one of the most rapidly expanding trends in onshore domestic oil and gas exploration and production today (Fig. 1 and 2). Shale gas is present in hydrocarbon rich shale formations. Shallow gas is commonly defined as gas occurrences in unconsolidated sediments of Tertiary age (often down to depths of 1000 m below surface). The occurrences are positively associated with thick Neogene sediments and are often trapped in anticlinal structures associated with rising salt domes (Muntendam-Bos et al, 2009). Shale has low matrix permeability, so gas production in commercial quantities requires fractures to provide permeability. -
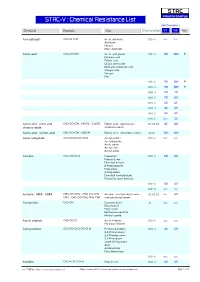
STAC-V : Chemical Resistance List Max Temperature
S TA C Industrial Coatings STAC-V : Chemical Resistance List Max Temperature Chemical Formula Alias Concentration V1 V2 Note Acetaldehyde CH3-CH=O Acetic aldehyde 100 % n.r. n.r. Aldehyde Ethanal Ethyl aldehyde Acetic acid CH3-CO-OH Acetic acid glacial 010 % 90 100 0 Ethanoic acid Ethylic acid Glacial acetic acid Methane carboxylic acid Vinegar acid Vinegar Hac 015 % 90 100 0 025 % 90 100 0 040 % 80 90 050 % 70 80 075 % 60 65 080 % 45 45 085 % 45 45 100 % n.r. 25 Acetic acid : nitric acid : CH3-CO-OH : HNO3 : Cr2O3 Ethylic acid : salpeterzuur : 03:05:03 65 80 chromic oxide chromium oxide Acetic acid : sulfuric acid CH3-CO-OH : H2SO4 Ethylic acid : dihydrogen sulfate 20:10 100 100 Acetic anhydride CH3-CO-O-CO-CH3 Acetyl acetate 100 % n.r. n.r. Acetanhydride Acetic oxide Acetyl ether Acetyl oxide Acetone CH3-CO-CH3 Propanone 005 % 80 80 Propan-2-one Dimethyl ketone β-Ketopropane[ Propanone 2-Propanone Dimethyl formaldehyde Pyroacetic spirit (archaic) 010 % 80 80 100 % n.r. n.r. Acetone : MEK : MiBK CH3-CO-CH3 : CH3-CO-CH2- Acetone : methylethyl ketone : 02:02:02 n.r. 40 CH3 : CH3-CO-CH2-CH2-CH3 methylisobutyl ketone Acetonitrile CH3-CN Cyanomethane all n.r. n.r. Ethanenitrile Ethyl nitrile Methanecarbonitrile Methyl cyanid Acetyl chloride CH3-CO-Cl Acetic chloride 100 % n.r. n.r. Ethanoyl chloride Acetylacetone CH3-CO-CH2-CO-CH3 Pentane-2,4-dione 020 % 40 50 2,4-Pentanedione 2,4-Dioxopentane 2,4-Pentadione acetyl-2-Propanone Acac Acetoacetone Diacetylmethane 100 % n.r. -

Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC
Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC R E S E A R C H A N D D E V E L O P M E N T EPA/600/R-06/105 September 2006 Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC By S. H. Hilal Ecosystems Research Division National Exposure Research Laboratory Athens, Georgia U.S. Environmental Protection Agency Office of Research and Development Washington, DC 20460 NOTICE The information in this document has been funded by the United States Environmental Protection Agency. It has been subjected to the Agency's peer and administrative review, and has been approved for publication. Mention of trade names of commercial products does not constitute endorsement or recommendation for use. ii ABSTRACT SPARC (SPARC Performs Automated Reasoning in Chemistry) chemical reactivity models were extended to calculate hydrolysis rate constants for carboxylic acid ester and phosphate ester compounds in aqueous non- aqueous and systems strictly from molecular structure. The energy differences between the initial state and the transition state for a molecule of interest are factored into internal and external mechanistic perturbation components. The internal perturbations quantify the interactions of the appended perturber (P) with the reaction center (C). These internal perturbations are factored into SPARC’s mechanistic components of electrostatic and resonance effects. External perturbations quantify the solute-solvent interactions (solvation energy) and are factored into H-bonding, field stabilization and steric effects. These models have been tested using 1471 reliable measured base, acid and general base-catalyzed carboxylic acid ester hydrolysis rate constants in water and in mixed solvent systems at different temperatures. -

Asymmetric Synthesis of Organophosphates and Their
ASYMMETRIC SYNTHESIS OF ORGANOPHOSPHATES AND THEIR DERIVATIVES Thesis Submitted to The College of Arts and Sciences of the UNIVERSITY OF DAYTON In Partial Fulfillment of the Requirements for The Degree of Master of Science in Chemistry By Batool J. Murtadha Dayton, Ohio May 2020 ASYMMETRIC SYNTHESIS OF ORGANOPHOSPHATES AND THEIR DERIVATIVES Name: Murtadha, Batool J. APPROVED BY: __________________________________ Jeremy Erb, Ph.D. Research Advisor Assistant Professor Department of Chemistry University of Dayton ___________________________________ Vladimir Benin, Ph.D. Professor of Chemistry Department of Chemistry University of Dayton ___________________________________ Justin C. Biffinger, Ph.D. Committee Member Assistant Professor Department of Chemistry University of Dayton ii © Copyright by Batool J. Murtadha All rights reserved 2020 iii ABSTRACT ASYMMETRIC SYNTHESIS OF ORGANOPHOSPHATES AND THEIR DERIVATIVES Name: Murtadha, Batool J. University of Dayton Advisor: Dr. Jeremy Erb Organophosphorus compounds (OPs) are widely used in the agricultural industry especially in the pesticide market. Phosphates play a huge role as biological compounds in the form of energy carrier compounds like ATP, and medicine as antivirals. OPs have become increasingly important as evidenced by the publication of new methods devoted to their uses and synthesis. These well-established studies lay the basis for industrial organic derivatives of phosphorus preparations. The current work explored methods of synthesizing chiral organophosphate triesters. We experimented with different processes roughly divided into either an electrophilic or nucleophilic strategy using chiral Lewis acids, organocatalysts (HyperBTM), activating agents, and chiral auxiliaries with the goal of control stereoselectivity. These methods were explored through the use of different starting materials like POCl3, triethyl phosphate, methyl phosphordichloradate, and PSCl3. -

I Norgan Ic C He Mi Str Y
View Article Online / Journal Homepage / Table of Contents for this issue INORGANIC CHEMISTRY. 443 I n o r g a n ic C h e mi s t r y. Composition of Atmospheres which Extinguish Flame. By FRANKCLOWES ( PTOC. Roy. Soc., 1894, 56, 2-6) .--The experimental flame burning at a platinum jet 1 mm. in diameter, was 0.75 in. in height; it was gradually lowered into a cylinder containing the atmosphere of mixed gases, and these were considered to be in extinctive proportions if the flame was extinguished during its downward passage, or immediately on attaining its lowest position in the cylinder. The gaseous mixture was regarded as containing the minimum quantitr of extinctive gas, when the flame on being lowered into another mixture containing 1 per cent. less of such gas continued to burn in it for a few seconds before being ex- tinguished. Experiments made with flames of hydrogen and Published on 01 January 1895. Downloaded 25/10/2014 07:12:09. alcohol, varying from 0.4 in. to 1.5 in. in height, show that the varying dimensions of the flame are without influence on the proportion of carbonic anhydride in the air necessary to pro- duce extinction. Characteristic differences were observed between the behaviour of wick-fed flames and that of gas-fed flames when they were introduced into an atmosphere which extinguished them, the wick-fed flames gradually diminishing in size until they vanished, whilst the gas-fed flames gradually increased in size, becoming paler and apparently lower in temperature until they suddenly expired. -

The Iron-Dependent Cyanide and Hydrogen Peroxide Co-Toxicity in Escherichia Coli and Its Catastrophic Consequences for the Chromosome
THE IRON-DEPENDENT CYANIDE AND HYDROGEN PEROXIDE CO-TOXICITY IN ESCHERICHIA COLI AND ITS CATASTROPHIC CONSEQUENCES FOR THE CHROMOSOME BY TULIP MAHASETH DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Microbiology in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Andrei Kuzminov, Chair and Director of Research Professor John E. Cronan Professor Jeffrey F. Gardner Associate Professor Carin K. Vanderpool ABSTRACT 2+ Hydrogen peroxide (H2O2) can oxidize cytoplasmic ferrous ions (Fe ) to produce highly reactive hydroxyl radicals (•OH) via Fenton’s reaction that can damage various biomolecules causing oxidative stress. Even though at concentrations higher than 20 mM H2O2 by itself can efficiently kill micro-organisms, it is metabolically impossible for eukaryotic cells to generate H2O2, an uncharged molecule, in such large quantities inside the cell. We propose that potentiation of physiologically relevant amounts of H2O2 by various small molecules serves as a more feasible and safe mechanism to combat invading microbes. NO potentiation of H2O2 toxicity is a known bactericidal weapon employed by macrophages. In fact, in human neutrophils activated by bacterial infection, the myeloperoxidase enzyme catalyzes the formation of hydrogen cyanide (HCN) from serum thiocyanate (SCN-). In the past, researchers have reported that a combination of low millimolar doses of H2O2 and cyanide (CN), which are individually bacteriostatic, caused rapid synergistic killing in Escherichia coli. Our aim is to understand the immune cells antimicrobial responses by investigating the mechanism of CN potentiation of H2O2 toxicity and its chromosomal consequences. We have found that the ability of CN to recruit iron from intracellular depots such as ferritin contributes to its potentiation of H2O2 toxicity, whereas the major stationary phase intracellular iron depot protein, Dps, can sequester this iron, thereby quelling Fenton's reaction. -

Synthesis of 4-Phosphono Β-Lactams and Related Azaheterocyclic Phosphonates
SYNTHESIS OF 4-PHOSPHONO β-LACTAMS AND RELATED AZAHETEROCYCLIC PHOSPHONATES IR . KRISTOF MOONEN To Elza Vercauteren Promotor: Prof. dr. ir. C. Stevens Department of Organic Chemistry, Research Group SynBioC Members of the Examination Committee: Prof. dr. ir. N. De Pauw (Chairman) Prof. dr. J. Marchand-Brynaert Prof. dr. A. Haemers Prof. dr. S. Van Calenbergh Prof. dr. ir. E. Vandamme Prof. dr. ir. R. Verhé Prof. dr. ir. N. De Kimpe Dean: Prof. dr. ir. H. Van Langenhove Rector: Prof. dr. P. Van Cauwenberge IR . KRISTOF MOONEN SYNTHESIS OF 4-PHOSPHONO β-LACTAMS AND RELATED AZAHETEROCYCLIC PHOSPHONATES Thesis submitted in fulfillment of the requirements for the degree of Doctor (PhD) in Applied Biological Sciences: Chemistry Dutch translation of the title: Synthese van 4-fosfono-β-lactamen en aanverwante azaheterocyclische fosfonaten ISBN-Number: 90-5989-129-5 The author and the promotor give the authorisation to consult and to copy parts of this work for personal use only. Every other use is subject to the copyright laws. Permission to reproduce any material contained in this work should be obtained from the author. Woord Vooraf Toen ik op een hete dag in de voorbije zomer dit woord vooraf schreef, stond ik voor één van de laatste horden te nemen in de weg naar het “doctoraat”. Het ideale moment voor een nostalgische terugblik op een zeer fijne periode, hoewel het onzinnig zou zijn te beweren dat alles rozegeur en maneschijn was. En op het einde van de rit komt dan ook het moment waarop je eindelijk een aantal mensen kunt bedanken, omwille van sterk uiteenlopende redenen. -

US 2004/0237384 A1 Orr (43) Pub
US 2004O237384A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0237384 A1 Orr (43) Pub. Date: Dec. 2, 2004 (54) FUEL COMPOSITIONS EXHIBITING (52) U.S. Cl. ................. 44/314; 44/320; 44/358; 44/359; IMPROVED FUEL STABILITY 44/360; 44/444 (76) Inventor: William C. Orr, Denver, CO (US) Correspondence Address: (57)57 ABSTRACT HOGAN & HARTSON LLP ONE TABOR CENTER, SUITE 1500 A fuel composition of the present invention exhibits mini 1200 SEVENTEENTH ST mized hydrolysis and increased fuel Stability, even after DENVER, CO 80202 (US) extended storage at 65 F. for 6–9 months. The composition, which is preferably not strongly alkaline (3.0 to 10.5), is (21) Appl. No.: 10/722,063 more preferably weakly alkaline to mildly acidic (4.5 to 8.5) (22) Filed: Nov. 24, 2003 and most preferably slightly acidic (6.3 to 6.8), includes a e ars lower dialkyl carbonate, a combustion improving amount of Related U.S. Application Data at least one high heating combustible compound containing at least one element Selected from the group consisting of (63) Continuation-in-part of application No. 08/986,891, aluminum, boron, bromine, bismuth, beryllium, calcium, filed on Dec. 8, 1997, now Pat. No. 6,652,608. cesium, chromium, cobalt, copper, francium, gallium, ger manium, iodine, iron, indium, lithium, magnesium, manga Publication Classification nese, molybdenum, nickel, niobium, nitrogen, phosphorus, potassium, palladium, rubidium, Sodium, tin, Zinc, (51) Int. Cl." ........ C10L 1/12; C1OL 1/30; C1OL 1/28; praseodymium, rhenium, Silicon, Vanadium, or mixture, and C1OL 1/18 a hydrocarbon base fuel. -

Bioorganic Chemistry
Hermann Dugas Christopher Penney Bioorganic Chemistry A Chemical Approach to Enzyme Action With 82 Figures Spri nger-Verlag New York Heidelberg Berlin Dr. Hermann Dugas Dr. Christopher Penney Departement de Chimie Connaught Research Institute Universite de Montreal Willowdale, Ontario Montreal, Quebec Canada M2N 5T8 Canada H3C 3Vl Series Editor: Prof. Charles R. Cantor Columbia University Box 608 Havemeyer Hall New York, New York 10027 USA Cover: The green illustration represents the hypothetical mode of binding of a rigid structural analogue of N-benzoyl-L-phenylalanine methyl ester at the active site of a-chymotrypsin. The illustration emphasizes the equilibration toward the favored configuration (see text page 224). The background design is taken from a diagrammatic representation of the primary structure of a-chymotrypsin. After Nature with permission [B.W. Matthews, P.B. Sigler, R. Henderson, and D.M. Blow (1967), Nature 214, 652-656]. Library of Congress Cataloging in Publication Data Dugas, Hermann, 1942- Bioorganic chemistry. (Springer advanced texts in chemistry.) Bibliography: p. Includes index. 1. Enzymes. 2. Biological chemistry. 3. Chemistry,. Organic. I. Penney, Christopher, 1950- joint author. II. Title. m. Series. [DNLM: 1. Biochemistry. 2. Enzymes-Metabolism. QUl35 D866b] QP60 1. D78 574.19'25 80-16222 All rights reserved. No part of this book may be translated or reproduced in any form without written permission from Springer-Verlag. The use of general descriptive names, trade names, trademarks, etc. in this publication, even if the former are not especially identified, is not to be taken as a sign that such names, as understood by the Trade Marks and Merchandise Marks Act, may accordingly be used freely by anyone. -

Division of Polymer Chemistry (POLY)
Division of Polymer Chemistry (POLY) Graphical Abstracts Submitted for the 258th ACS National Meeting & Exposition August 25 - 29, 2019 | San Diego, CA Table of Contents [click on a session time (AM/PM/EVE) for link to abstracts] Session SUN MON TUE WED THU AM AM Polymerization-Induced Nanostructural Transitions PM PM Paul Flory's "Statistical Mechanics of Chain Molecules: The 50th AM AM Anniversary of Polymer Chemistry" PM PM AM AM AM Eco-Friendly Polymerization PM PM EVE AM Characterization of Plastics in Aquatic Environments PM PM AM General Topics: New Synthesis & Characterization of Polymers AM PM AM PM EVE Future of Biomacromolecules at a Crossroads of Polymer Science & AM AM EVE Biology PM PM Industrial Innovations in Polymer Science PM AM AM Polymers for Defense Applications PM AM PM EVE Henkel Outstanding Graduate Research in Polymer Chemistry in AM Honor of Jovan Kamcev AM AM Polymeric Materials for Water Purification PM AM PM EVE Young Industrial Polymer Scientist Award in Honor of Jason Roland AM Biomacromolecules/Macromolecules Young Investigator Award PM Herman F. Mark Award in Honor of Nicholas Peppas AM DSM Graduate Student Award AM Overberger International Prize in Honor of Kenneth Wagner PM Note: ACS does not own copyrights to the individual abstracts. For permission, please contact the author(s) of the abstract. POLY 1: High throughput and solution phase TEM for discovery of new pisa reaction manifolds Nathan C. Gianneschi1, [email protected], Mollie A. Touve1, Adrian Figg1, Daniel Wright1, Chiwoo Park2, Joshua Cantlon3, Brent S. Sumerlin4. (1) Chemistry, Northwestern University, Evanston, Illinois, United States (2) Florida State University, Tallahassee, Florida, United States (3) SCIENION, San Francisco, California, United States (4) Department of Chemistry, University of Florida, Gainesville, Florida, United States We describe the development of a high-throughput, automated method for conducting TEM characterization of materials, to remove this bottleneck from the discovery process.