Bioorganic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Courses 2005-2006
Chemistry Courses 2005-2006 Autumn 2005 Chem 11101 General Chemistry I, Variant A Lee Chem 11102 General Chemistry I, Variant B Norris Chem 12200 Honors General Chemistry I Levy Chem 22000 Organic Chemistry I Yu Chem 22300 Intermediate Organic Chemistry Mrksich Chem 26100 Quantum Mechanics Mazziotti Chem 30100 Advanced Inorganic Chemistry Hopkins Chem 30900 Bioinorganic Chemistry He Chem 32100 Physical Organic Chemistry I Ismagilov Chem 32200 Organic Synthesis and Structure Rawal Chem 32600 Protein Fundamentals Piccirilli Chem 36100 Wave Mechanics & Spectroscopy Butler Chem 36400 Chemical Thermodynamics Dinner Winter 2006 Chem 11201 General Chemistry II, Variant A Scherer Chem 11202 General Chemistry II, Variant B Butler Chem 12300 Honors General Chemistry II Dinner Chem 20100 Inorganic Chemistry I Hillhouse Chem 22100 Organic Chemistry II Rawal Chem 23100 Honors Organic Chemistry II Kozmin Chem 26200 Thermodynamics Norris Chem 26700 Experimental Physical Chemistry Levy Chem 30200 Synthesis & Physical Methods in Inorganic Chemistry Jordan Chem 30400 Organometallic Chemistry Bosnich Chem 32300 Tactics of Organic Synthesis Yamamoto Chem 32400 Physical Organic Chemistry II Mrksich Chem 36200 Quantum Mechanics Freed Chem 36300 Statistical Mechanics Mazziotti Chem 38700 Biophysical Chemistry Lee Spring 2006 Chem 11301 General Chemistry III, Variant A Kozmin Chem 11302 General Chemistry III, Variant B Guyot-Sionnest Chem 20200 Inorganic Chemistry II Jordan Chem 22200 Organic Chemistry III Kent Chem 23200 Honors Organic Chemistry III Yamamoto Chem 22700 Advanced Organic / Inorganic Laboratory (8 students) He Chem 26300 Chemical Kinetics and Dynamics Sibner Chem 26800 Computational Chemistry & Biology Freed Chem 30600 Chemistry of the Elements Hillhouse Chem 31100 Supramolecular Chemistry Bosnich Chem 32500 Bioorganic Chemistry Piccirilli Chem 32900 Polymer Chemistry Yu Chem 33000 Complex Systems Ismagilov Chem 36500 Chemical Dynamics Scherer Chem 36800 Advanced Computational Chemistry & Biology Freed . -

Curriculum of Department of Pharmacy (6-Year Program)
Curriculum of Department of Pharmacy (6-year program) 1st year 2nd year 3rd year 4th year 5th year 6th year Basic Subjects for Pharmacy Student; Lectures and Practices of Basic Specialized Lectures and Practices Drug Therapy and its Related Practical Training and Beginning of Research for Graduation and SGL and more Sciences Research for Graduation Advanced Lectures Fundamental Education Professional Education Ⅰ Professional Education Ⅱ ■Humanism ■Humanism ■Preparatory Pharmacy Education ■Professional Pharmacy Education ■Practical Training ■Advanced Education Introduction to Humanism I Introduction to Humanism Ⅱ English for Pharmacy Oriental Medicine Preparation for Practical Training Research for Graduation Pharmacotherapeutics Practical Training in Hospital Advanced Clinical Training ■Preparatory Pharmacy Education ■Professional Pharmacy Education Drug Information Science Practical Training in Community Phrarmacy General Pharmacy Practice II ■Introduction of Pharmacy Basic English and English for Pharmacy Clinical Chemistry Drug Development and Production Japanese Traditional Medicine Invitation to Pharmacy Basic Statistics Synthesis of Target Molecules Pharmacy and Society ■Advanced Education Clinical Pharmacokinetics Early Exposure to Pharmacy Practical Trainings for Pharmacy Bioorganic Chemistry General Pharmacy Practice Research for Graduation Clinical Drug Evaluation Students Pharmaceutical Health Care Training Medical Economy Natural Products Chemistry ■Practical Training Education Drug Safty Evaluation and Pharmacoepidemiology -

Recreating Ancient Metabolic Pathways Before Enzymes Kamila B
Recreating ancient metabolic pathways before enzymes Kamila B. Muchowska, Elodie Chevallot-Beroux, Joseph Moran To cite this version: Kamila B. Muchowska, Elodie Chevallot-Beroux, Joseph Moran. Recreating ancient metabolic path- ways before enzymes. Bioorganic and Medicinal Chemistry, Elsevier, 2019, 27 (12), pp.2292-2297. 10.1016/j.bmc.2019.03.012. hal-02516541 HAL Id: hal-02516541 https://hal.archives-ouvertes.fr/hal-02516541 Submitted on 23 Mar 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Graphical Abstract To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered. Recreating ancient metabolic pathways Leave this area blank for abstract info. before enzymes Kamila B. Muchowskaa* , Elodie Chevallot-Berouxa, and Joseph Morana* University of Strasbourg, CNRS, ISIS UMR 7006, 67000 Strasbourg, France Bioorganic & Medicinal Chemistry journal homepage: www.elsevier.com Recreating ancient metabolic pathways before enzymes Kamila B. Muchowska,a* Elodie Chevallot-Beroux,a and Joseph Morana* a University of Strasbourg, CNRS, ISIS UMR 7006, 67000 Strasbourg, France. ARTICLE INFO ABSTRACT Article history: The biochemistry of all living organisms uses complex, enzyme-catalyzed metabolic reaction Received networks. -

Shale Gas and Groundwater Quality
Shale Gas and Groundwater Quality A literature review on fate and effects of added chemicals Alette Langenhoff 1202141-008 © Deltares, 2011 1202141-008-ZWS-0001, 28 December 2011, final Contents 1 Introduction 1 2 The process of fracturing or fracking 5 3 The use of chemicals 7 4 Polyacrylamide 8 4.1 Aerobic degradation of polyacrylamide 8 4.2 Anaerobic degradation 10 4.3 Chemical or physical removal 10 4.4 Conclusion on removal of polyacrylamide 10 5 Glutaraldehyde 12 5.1 Biocide 12 5.2 Biodegradation 12 5.3 Chemical inactivation of glutaraldehyde 13 6 Conclusions 14 7 References 15 Appendices 17 Appendices A Chemicals identified in hydraulic fracturing fluid and flowback/produced water (EPA, 2011). A-1 B Fracturing fluid ingredients and common uses (Europe Unconventional Gas 2011) B-1 C Properties of Polyacrylamide (source: Wikipedia) C-1 D Properties of Glutaraldehyde (source: Wikipedia) D-1 Shale Gas and Groundwater Quality i 1202141-008-ZWS-0001, 28 December 2011, final 1 Introduction Shale gas is a so-called unconventional sources of natural gas, and is one of the most rapidly expanding trends in onshore domestic oil and gas exploration and production today (Fig. 1 and 2). Shale gas is present in hydrocarbon rich shale formations. Shallow gas is commonly defined as gas occurrences in unconsolidated sediments of Tertiary age (often down to depths of 1000 m below surface). The occurrences are positively associated with thick Neogene sediments and are often trapped in anticlinal structures associated with rising salt domes (Muntendam-Bos et al, 2009). Shale has low matrix permeability, so gas production in commercial quantities requires fractures to provide permeability. -
STAC-V : Chemical Resistance List Max Temperature
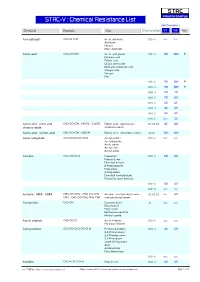
S TA C Industrial Coatings STAC-V : Chemical Resistance List Max Temperature Chemical Formula Alias Concentration V1 V2 Note Acetaldehyde CH3-CH=O Acetic aldehyde 100 % n.r. n.r. Aldehyde Ethanal Ethyl aldehyde Acetic acid CH3-CO-OH Acetic acid glacial 010 % 90 100 0 Ethanoic acid Ethylic acid Glacial acetic acid Methane carboxylic acid Vinegar acid Vinegar Hac 015 % 90 100 0 025 % 90 100 0 040 % 80 90 050 % 70 80 075 % 60 65 080 % 45 45 085 % 45 45 100 % n.r. 25 Acetic acid : nitric acid : CH3-CO-OH : HNO3 : Cr2O3 Ethylic acid : salpeterzuur : 03:05:03 65 80 chromic oxide chromium oxide Acetic acid : sulfuric acid CH3-CO-OH : H2SO4 Ethylic acid : dihydrogen sulfate 20:10 100 100 Acetic anhydride CH3-CO-O-CO-CH3 Acetyl acetate 100 % n.r. n.r. Acetanhydride Acetic oxide Acetyl ether Acetyl oxide Acetone CH3-CO-CH3 Propanone 005 % 80 80 Propan-2-one Dimethyl ketone β-Ketopropane[ Propanone 2-Propanone Dimethyl formaldehyde Pyroacetic spirit (archaic) 010 % 80 80 100 % n.r. n.r. Acetone : MEK : MiBK CH3-CO-CH3 : CH3-CO-CH2- Acetone : methylethyl ketone : 02:02:02 n.r. 40 CH3 : CH3-CO-CH2-CH2-CH3 methylisobutyl ketone Acetonitrile CH3-CN Cyanomethane all n.r. n.r. Ethanenitrile Ethyl nitrile Methanecarbonitrile Methyl cyanid Acetyl chloride CH3-CO-Cl Acetic chloride 100 % n.r. n.r. Ethanoyl chloride Acetylacetone CH3-CO-CH2-CO-CH3 Pentane-2,4-dione 020 % 40 50 2,4-Pentanedione 2,4-Dioxopentane 2,4-Pentadione acetyl-2-Propanone Acac Acetoacetone Diacetylmethane 100 % n.r. -

A Survey of Carbon Fixation Pathways Through a Quantitative Lens
Journal of Experimental Botany, Vol. 63, No. 6, pp. 2325–2342, 2012 doi:10.1093/jxb/err417 Advance Access publication 26 December, 2011 REVIEW PAPER A survey of carbon fixation pathways through a quantitative lens Arren Bar-Even, Elad Noor and Ron Milo* Department of Plant Sciences, The Weizmann Institute of Science, Rehovot 76100, Israel * To whom correspondence should be addressed. E-mail: [email protected] Received 15 August 2011; Revised 4 November 2011; Accepted 8 November 2011 Downloaded from Abstract While the reductive pentose phosphate cycle is responsible for the fixation of most of the carbon in the biosphere, it http://jxb.oxfordjournals.org/ has several natural substitutes. In fact, due to the characterization of three new carbon fixation pathways in the last decade, the diversity of known metabolic solutions for autotrophic growth has doubled. In this review, the different pathways are analysed and compared according to various criteria, trying to connect each of the different metabolic alternatives to suitable environments or metabolic goals. The different roles of carbon fixation are discussed; in addition to sustaining autotrophic growth it can also be used for energy conservation and as an electron sink for the recycling of reduced electron carriers. Our main focus in this review is on thermodynamic and kinetic aspects, including thermodynamically challenging reactions, the ATP requirement of each pathway, energetic constraints on carbon fixation, and factors that are expected to limit the rate of the pathways. Finally, possible metabolic structures at Weizmann Institute of Science on July 3, 2016 of yet unknown carbon fixation pathways are suggested and discussed. -

Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC
Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC R E S E A R C H A N D D E V E L O P M E N T EPA/600/R-06/105 September 2006 Estimation of Hydrolysis Rate Constants of Carboxylic Acid Ester and Phosphate Ester Compounds in Aqueous Systems from Molecular Structure by SPARC By S. H. Hilal Ecosystems Research Division National Exposure Research Laboratory Athens, Georgia U.S. Environmental Protection Agency Office of Research and Development Washington, DC 20460 NOTICE The information in this document has been funded by the United States Environmental Protection Agency. It has been subjected to the Agency's peer and administrative review, and has been approved for publication. Mention of trade names of commercial products does not constitute endorsement or recommendation for use. ii ABSTRACT SPARC (SPARC Performs Automated Reasoning in Chemistry) chemical reactivity models were extended to calculate hydrolysis rate constants for carboxylic acid ester and phosphate ester compounds in aqueous non- aqueous and systems strictly from molecular structure. The energy differences between the initial state and the transition state for a molecule of interest are factored into internal and external mechanistic perturbation components. The internal perturbations quantify the interactions of the appended perturber (P) with the reaction center (C). These internal perturbations are factored into SPARC’s mechanistic components of electrostatic and resonance effects. External perturbations quantify the solute-solvent interactions (solvation energy) and are factored into H-bonding, field stabilization and steric effects. These models have been tested using 1471 reliable measured base, acid and general base-catalyzed carboxylic acid ester hydrolysis rate constants in water and in mixed solvent systems at different temperatures. -

Editorial Highlights for Organi Chemistry: Current Research
n ISSN: 2161-0401 Organic Chemistry: Current Research Editorial Editorial Note on Bioorganic Chemistry Sandhya Kille Department of Microbiology, Acharya Nagarjuna University, India EDITORIAL structure-function studies of therapeutics, and bio conjugates. Students focused on this area at Oregon Bioorganic chemistry could also be a rapidly growing develop a solid foundation in synthetic chemistry and even content that mixes science and biochemistry. It’s that have the good thing about the outstanding branch of bioscience that deals with the study of biochemistry/molecular biology research environment at biological processes using chemical methods. UO. Bioorganic chemistry applies the principles and Protein and enzyme function are samples of those techniques of chemistry to unravel problems of biological processes. Sometimes biochemistry is utilized relevance, taking inspiration from biology to develop new interchangeably for bioorganic chemistry; the excellence chemical processes. being that bioorganic chemistry is chemistry that's Cornell bioorganic chemists emphasize chemical and focused on the biological aspects. While biochemistry molecular approaches to solving important biological aims at understanding biological processes using problems. Research areas include the applying of chemistry, bioorganic chemistry attempts to expand synthetic and scientific discipline to the study of enzymes, organic-chemical researches (that is, structures, metabolic pathways and nucleic acids. This includes the synthesis, and kinetics) toward biology. event of mechanism-based enzyme inhibitors; elucidation When investigating metalloenzymes and cofactors, of enzyme mechanism and structure and studies of bioorganic chemistry overlaps bioinorganic chemistry. coenzyme reactivity. Chemical investigations are extended Bioorganic Chemistry and Chemical Biology, broadly to studies of receptor recognition, hormone and drug defined, are fields during which organic synthetic activity, and also the mechanism of chemical chemistry plays an unlimited role within the life sciences. -

Laboratory Classes in Bioorganic Chemistry
MINISTRY OF HEALTH OF REPUBLIC OF BELARUS VITEBSK STATE MEDICAL UNIVERSITY LABORATORY CLASSES IN BIOORGANIC CHEMISTRY L.G. Hidranovich, O.A. Khodos ( 2 - e « З Д / ) For Foreign students of the 1-st year Vitebsk 20t£ УДК 54 (042.3/4) ББК 24.239 L.G. Hidranovich, О.Л. Khodos LABORATORY CLASSES IN BIOORGANIC CHEMISTRY for foreign students of the 1-sl year: Manual./ L.G. Hidranovich, O.A. Khodos. - Vitebsk: v s m u , 20i b - 128 p. ( 2 - е м а д д ISBN 978-985-466-5$S‘-3 This issue contains program questions, problems, laboratory' works for the classes in bioorgamc chemistry, examination questions, tests, reference tables.The issue was wrote according to the typical educational program for the students o f higher medical educa tional establishments. Утверждено и рекомендовано к изданию Центральным учебно-научно методическим Советом непрерывного медицинского и фармацевтического образо вания Витебского государственного медицинского университета, 21.04________ 2007 г , протокол №4. ISBN 978-985-466-581-3 У Д К 54 (042.3/4) ББК 24.239 © Гидранович Л.Г., Ходос О А., 20/3 ©УО «Витебский государственный медицинский университет», 20'Й CONTENTS Thematic p!a:: of the lectures. 4 Thematic plan of the laboratory classes. 5 Accident prevention. 6 Theme 1. Classification and FJPAC nomenclature of organic com 7 pounds. Theme 2. Electronic structure of chemical bonds. Inductive and reso 9 nance effects. Theme 3. Stereochemistry of organic compounds. 11 Configuration and conformation of the organic compounds. Theme 4 Acid-base properties of organic compounds. 14 Theme 5. Classification and mechanisms of the reactions in organic 16 chemistry Saturated, unsaturated and aromatic hydrocarbons. -

I Norgan Ic C He Mi Str Y
View Article Online / Journal Homepage / Table of Contents for this issue INORGANIC CHEMISTRY. 443 I n o r g a n ic C h e mi s t r y. Composition of Atmospheres which Extinguish Flame. By FRANKCLOWES ( PTOC. Roy. Soc., 1894, 56, 2-6) .--The experimental flame burning at a platinum jet 1 mm. in diameter, was 0.75 in. in height; it was gradually lowered into a cylinder containing the atmosphere of mixed gases, and these were considered to be in extinctive proportions if the flame was extinguished during its downward passage, or immediately on attaining its lowest position in the cylinder. The gaseous mixture was regarded as containing the minimum quantitr of extinctive gas, when the flame on being lowered into another mixture containing 1 per cent. less of such gas continued to burn in it for a few seconds before being ex- tinguished. Experiments made with flames of hydrogen and Published on 01 January 1895. Downloaded 25/10/2014 07:12:09. alcohol, varying from 0.4 in. to 1.5 in. in height, show that the varying dimensions of the flame are without influence on the proportion of carbonic anhydride in the air necessary to pro- duce extinction. Characteristic differences were observed between the behaviour of wick-fed flames and that of gas-fed flames when they were introduced into an atmosphere which extinguished them, the wick-fed flames gradually diminishing in size until they vanished, whilst the gas-fed flames gradually increased in size, becoming paler and apparently lower in temperature until they suddenly expired. -

The Iron-Dependent Cyanide and Hydrogen Peroxide Co-Toxicity in Escherichia Coli and Its Catastrophic Consequences for the Chromosome
THE IRON-DEPENDENT CYANIDE AND HYDROGEN PEROXIDE CO-TOXICITY IN ESCHERICHIA COLI AND ITS CATASTROPHIC CONSEQUENCES FOR THE CHROMOSOME BY TULIP MAHASETH DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Microbiology in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Andrei Kuzminov, Chair and Director of Research Professor John E. Cronan Professor Jeffrey F. Gardner Associate Professor Carin K. Vanderpool ABSTRACT 2+ Hydrogen peroxide (H2O2) can oxidize cytoplasmic ferrous ions (Fe ) to produce highly reactive hydroxyl radicals (•OH) via Fenton’s reaction that can damage various biomolecules causing oxidative stress. Even though at concentrations higher than 20 mM H2O2 by itself can efficiently kill micro-organisms, it is metabolically impossible for eukaryotic cells to generate H2O2, an uncharged molecule, in such large quantities inside the cell. We propose that potentiation of physiologically relevant amounts of H2O2 by various small molecules serves as a more feasible and safe mechanism to combat invading microbes. NO potentiation of H2O2 toxicity is a known bactericidal weapon employed by macrophages. In fact, in human neutrophils activated by bacterial infection, the myeloperoxidase enzyme catalyzes the formation of hydrogen cyanide (HCN) from serum thiocyanate (SCN-). In the past, researchers have reported that a combination of low millimolar doses of H2O2 and cyanide (CN), which are individually bacteriostatic, caused rapid synergistic killing in Escherichia coli. Our aim is to understand the immune cells antimicrobial responses by investigating the mechanism of CN potentiation of H2O2 toxicity and its chromosomal consequences. We have found that the ability of CN to recruit iron from intracellular depots such as ferritin contributes to its potentiation of H2O2 toxicity, whereas the major stationary phase intracellular iron depot protein, Dps, can sequester this iron, thereby quelling Fenton's reaction. -

Bioorganic Chemistry
Lecture 1. Biomedical and Bioorganic Chemistry Lecturer Yanovska Anna Olexandrivna Calculate grade: • Total (200) • General module (80 total) • Tests (Nomenclature - 10 points, Heterofunctional compounds – 14 points, Lipids – 14 points, Aminoacids, Peptides – 14 points). • Individual homework (30 possible) • 20 points (laboratory works) • 18 points work in class • Grades: • 170-200 - excellent • 169 – 140 - good • 139 – 120 – satisfied • Less than 120 - unsatisfied Organic chemistry is the chemistry of compounds of carbon. Bioorganic chemistry is the part of organic chemistry that studies the carbon compounds, which are present in the living organism – the so-called biomolecules. The major biomolecules are carbohydrates, proteins, lipids, and nucleic acids. It also studies drugs and their derivatives. By carbon chain organic compounds are classified in following way: Organic Chemistry • The chemistry of carbon compounds. • What’s special about carbon? – tetravalent (sp3 hybridization) – wide choice in oxidation states – CO2 C, +4 – CH4 C, -4 – bonds well to O, N, halides,itself,etc. – Covalent bonds are very strong Bond formation in molecules of organic compounds Functional Groups Term used to refer to parts of organic molecules where reactions tend to occur. By amount of functional groups and their type organic compounds are classified onto - Monofunctional (one functional group): alcohol C2H5-OH, carboxylic acid CH3COOH - Polyfunctional – has two or more same functional groups: HO-CH2-CH2-OH - Heterofunctional – has two or more different functional groups: HO-CH2-CH2-NH2 Functional Group is a part of an organic compound, by which it belongs to class of organic compounds and has specific properties. Structure of Carbon Compounds • There are three hybridization states and geometries found in organic compounds: – sp3 Tetrahedral – sp2 Trigonal planar – sp Linear Hydrocarbons (contain only H and C) • Four types: – Alkanes – Alkenes – Alkynes – Aromatic hydrocarbon s Alkanes • Only single bonds.