Open Thesis Master Document V5.0.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -
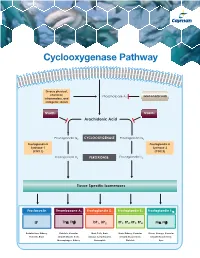
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

The Protective Effects of Conjugated Linoleic Acid Against Carcinogenesis
The Protective Effects of Conjugated Linoleic Acid Against Carcinogenesis Item Type text; Electronic Dissertation Authors Kemp, Michael Quentin Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 24/09/2021 15:13:15 Link to Item http://hdl.handle.net/10150/193637 THE PROTECTIVE EFFECTS OF CONJUGATED LINOLEIC ACID AGAINST CARCINOGENESIS by Michael Quentin Kemp A Dissertation Submitted to the Faculty of the GRADUATE PROGRAM IN NUTRITIONAL SCIENCES In Partial Fulfillment of the Requirements for the Degree of DOC TORAL OF PHILOSOPHY In the Graduate College THE UNIVERSITY OF ARIZONA 2005 2 THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE As members of the Dissertation Committee, we certify that we have read the dissertation prepared by Michael Q. Kemp entitled The Protective Effec ts of Conjugated Linoleic Acid Against Carcinogenesis and recommend that it be accepted as fulfilling the dissertation requirement for the Deg ree of Doctor of Philosophy _______ ___________________________ __________________ ________ _______ ___ _ Date : 11/18/05 . Donato Romagnolo _______ ___________________________ __________________ ________ ___________ Date: 11/18/05 . Wanda Howell _______ ___________________________ __________________ ________ ___________ Date: 11/18/05 . Linda Houtkooper _______ ___________________________ __________________ ________ ___________ Date: 11/18/05 . Scott Going _______ ___________________________ __________________ ________ ___________ Date: 11/18/05 . Joy Winzerling Final approval and acceptance of this dissertation is contingent upon the candidate’s submission of the final copies of the dissertation to the Graduate College. -

Regulation of Tissue Inflammation by 12-Lipoxygenases
biomolecules Review Regulation of Tissue Inflammation by 12-Lipoxygenases Abhishek Kulkarni 1 , Jerry L. Nadler 2, Raghavendra G. Mirmira 1,* and Isabel Casimiro 1,* 1 Department of Medicine, The University of Chicago, Chicago, IL 60637, USA; [email protected] 2 Department of Medicine and Pharmacology, New York Medical College, Valhalla, NY 10595, USA; [email protected] * Correspondence: [email protected] (R.G.M.); [email protected] (I.C.) Abstract: Lipoxygenases (LOXs) are lipid metabolizing enzymes that catalyze the di-oxygenation of polyunsaturated fatty acids to generate active eicosanoid products. 12-lipoxygenases (12-LOXs) primarily oxygenate the 12th carbon of its substrates. Many studies have demonstrated that 12-LOXs and their eicosanoid metabolite 12-hydroxyeicosatetraenoate (12-HETE), have significant pathological implications in inflammatory diseases. Increased level of 12-LOX activity promotes stress (both oxidative and endoplasmic reticulum)-mediated inflammation, leading to damage in these tissues. 12-LOXs are also associated with enhanced cellular migration of immune cells—a characteristic of several metabolic and autoimmune disorders. Genetic depletion or pharmacological inhibition of the enzyme in animal models of various diseases has shown to be protective against disease development and/or progression in animal models in the setting of diabetes, pulmonary, cardiovascular, and metabolic disease, suggesting a translational potential of targeting the enzyme for the treatment of several disorders. In this article, we review the role of 12-LOXs in the pathogenesis of several diseases in which chronic inflammation plays an underlying role. Citation: Kulkarni, A.; Nadler, J.L.; Keywords: 12-lipoxygenases; 12-LOXs; 12/15-lipoxygenase; 12/15-LOX; lipoxygenases; eicosanoids; Mirmira, R.G.; Casimiro, I. -

Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase Is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Spring 2010 Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Michelle Kinder University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Immunology and Infectious Disease Commons Recommended Citation Kinder, Michelle, "Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis" (2010). Publicly Accessible Penn Dissertations. 88. https://repository.upenn.edu/edissertations/88 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/88 For more information, please contact [email protected]. Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Abstract Fatty acid metabolism governs critical cellular processes in multiple cell types. The goal of my dissertation was to investigate the intersection between fatty acid metabolism and hematopoiesis. Although fatty acid metabolism has been extensively studied in mature hematopoietic subsets during inflammation, in developing hematopoietic cells the role of fatty acid metabolism, in particular by 12/ 15-Lipoxygenase (12/15-LOX), was unknown. The observation that 12/15-LOX-deficient (Alox15) mice developed a myeloid leukemia instigated my studies since leukemias are often a consequence of dysregulated hematopoiesis. This observation lead to the central hypothesis of this dissertation which is that polyunsaturated fatty acid metabolism mediated by 12/15-LOX participates in hematopoietic development. Using genetic mouse models and in vitro and in vivo cell development assays, I found that 12/15-LOX indeed regulates multiple stages of hematopoiesis including the function of hematopoietic stem cells (HSC) and the differentiation of B cells, T cells, basophils, granulocytes and monocytes. -

University of Groningen Reflections on Flurbiprofen Eyedrops Van Sorge
University of Groningen Reflections on flurbiprofen eyedrops van Sorge, Adriaan Alastair IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2002 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): van Sorge, A. A. (2002). Reflections on flurbiprofen eyedrops. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 25-09-2021 REFLECTIONS ON FLURBIPROFEN EYEDROPS REFLECTIONS ON FLURBIPROFEN EYEDROPS RIJKSUNIVERSITEIT GRONINGEN REFLECTIONS ON FLURBIPROFEN EYEDROPS REFLECTIONS ON FLURBIPROFEN EYEDROPS PROEFSCHRIFT ter verkrijging van het doctoraat in de Wiskunde en Natuurwetenschappen aan de Rijksuniversiteit Groningen, op gezag van de Rector Magnificus, dr. F. Zwarts, in het openbaar te verdedigen op maandag 2 december 2002 om 14.15 uur door Adriaan Alastair van Sorge geboren op 28 oktober 1944 te New Rochelle, New York, USA PROMOTORES Prof. -

Lipoxygenase Metabolites of Arachidonic Acid in Neuronal
TiPS - September 1990 [Vol. 11] 367 28 Takeda, N. et aL (1986) Acta Otolaryngol. Venerol. (Suppl. 115), 1-43 34 Pipkom, U. et al. (1987) Allergy 101,416-421 32 Granerus, G., Olafsson, I. H. and (Copenhagen) 42, 496-501 29 Tung, A. S. et al. (1985) Biochem. Roupe, G. (1985) Agents Actions 16, Pharmacol. 34, 3509-3515 244-248 30 August, T. F. el al. (1985) J. Pharm. Sci. 33 Neitaanmaki, H., Fraki, ]. E., Harvima, IPDllS1T: dimethyl-2-[4-(3-ethoxy-2- 74, 871-875 R. J. and Forstrom, L. (1989) Arch. hydroxypropoxy)phenylcarbamoyllethyl 31 Olafsson, I. H. (I985) Acta Derm. Dermatol. Res. 281, 99-104 sulfonium-p-toluene sulfonate polyacrylamide gel electrophor- Lipoxygenase metabolites of esis) and has no apparent cofactor requirement 4. A cDNA encoding arachidonic acid in neuronal leukocyte 12-1ipoxygenase has been isolated and sequenced 5. Like all other lipoxygenases, 12- transmembrane signalling lipoxygenase catalyses the intro- duction of molecular oxygen into Daniele Piomelli and Paul Greengard a 1,4-(cis,cis)-pentadiene moiety, converting arachidonic acid into the hydroperoxide, (12s)-hydro- Studies of invertebrate and vertebrate nervous tissue have demonstrated that peroxyeicosatetraenoic acid (12- free arachidonic acid and its lipoxygenase metabolites are produced in a HPETE) - a reaction that is both receptor-dependent fashion. The intracellular actions of these compounds regiospecific and stereospecific. include the regulation of activity of membrane ion channels and protein Other cis-polyunsaturated fatty kinases. In this article Daniele Piomelli and Paul Greengard review the acids, such as linoleic acid, lino- evidence that these lipophilic molecules constitute a novel class of intracellular lenic acid and docosahexaenoic second messenger, possibly involved in the modulation of neurotransmitter acid are also good substrates for release. -

Isolation and Structure of Two Prostaglandin Endoperoxides That
Proc. Nat. Acad. Sci. USA Vol. 71, No. 2, pp. 345-349, February 1974 Isolation and Structure of Two Prostaglandin Endoperoxides That Cause Platelet Aggregation (15-hydroperoxy endoperoxide/15-hydroxy endoperoxide/platelet aggregation/ contraction of rabbit aorta) MATS HAMBERG, JAN SVENSSON, TOSHIO WAKABAYASHI, AND BENGT SAMUELSSON Department of Chemistry, Karolinska Institutet, S 104 01, Stockholm, Sweden Communicated by Hugo Theorell, September 19, 1978 ABSTRACT Incubation for a short time of arachidonic (kindly provided by Dr. W. Stoffel, Cologne, Germany, see acid with the microsomal fraction of a homogenate of the ref. 2) and Na14CN followed by hydrolysis of the nitrile. The vesicular gland of sheep in the presence of 1 mM p-mer- curibenzoate followed by extraction and silicic acid chemical and radiochemical purity was in excess of 98%, chromatography yielded two prostaglandin endoper- as judged by thin-layer radiochromatography. Part of the oxides. The structures of these compounds, i.e., 15-hy- labeled acid was diluted with unlabeled material to make a droperoxy-9a,11a-peroxidoprosta-5,13-dienoic acid (pros- preparation with specific radioactivity of 0.77 Ci/mol, which taglandin G2) and 15-hydroxy-9a,lla-peroxidoprosta- 5,13-dienoic acid (prostaglandin H2), were assigned mainly was used for incubations with vesicular gland microsomes. by a number of chemical transformations into previously Thin-Layer Chromatography (TLC) was carried out with known prostaglandins. The new prostaglandins were 50- 200 times (prostaglandin G2) and 100-450 times (prosta- plates coated with chloroform-methanol-washed Silica gel G glandin H2) more active than prostaglandin E2 on the super- and the following solvent systems (when not otherwise fused aorta strip. -

Lipoxygenase-Generated Icosanoids Inhibit Glucose-Induced Insulin Release from Rat Islets
RostagiamJins Leukotrienes and Essential Fatty Acids (1990) 40.214 @ Longman Group UK Ltd 1990 Lipoxygenase-Generated Icosanoids Inhibit Glucose-induced Insulin Release from Rat Islets M. H. Nathan* and S. Belbez Pekt *Department of Internal Medicine (Division of Endocrinology and Metabolism), University of Michigan, Ann Arbor, Michigan, USA and ‘5560 Medical Sciences Research Building-2, University of Michigan Medical Center, Ann Arbor, MI 48109-0678, USA (Correspondence to SBP) ABSTRACT. Lipoxygenase-pathway metabolites of arachidonic acid are produced in pancreatic islets. They are are implicated in insulin release, since nonselective inhibitors of lipoxygenases inhibit glucose-induced insulin release. We studied the interplay in insulin release between glucose and selected icosanoids formed in 5-, 12- and 15-lipoxygenase pathways. Effects on immunoreactive insulin release of 10’ to 1O-6 12-(R)-HETE, 12-(S)-HETE, hepoxilin As, lipoxin Bq, LTB4 or LTC4 were tested individually in 30-min incubations of freshly isolated young adult Wistar rat pancreatic islets, in the presence of 5.6 mM or 23 mM glucose. Basal insulin release (at 5.6 mM glucose) was stimulated by LTC4 and hepoxilin A3 (304% and 234% of controls at 5.6 mM glucose alone, respectively), inhibited by 12-(S)-HPETE (56%), and was not affected by 12-(R)-HETE, 12-(S)-HETE, lipoxin B4 or LTB4 (ill%, 105%, 106% and 136%, respectively). Insulin release evoked by 23 mM glucose (190-320%) was inhibited (50-145%) by all icosanoids tested, except LTC4 (162%). We conclude that, among the lipoxygenase products tested, only leukotrienes and hepoxilin are candidates for a tonic-stimulatory influence on basal insulin release. -

NS-398 Upregulates Constitutive Cyclooxygenase-2 Expression in the M-1 Cortical Collecting Duct Cell Line
ARTICLES J Am Soc Nephrol 10: 2261–2271, 1999 NS-398 Upregulates Constitutive Cyclooxygenase-2 Expression in the M-1 Cortical Collecting Duct Cell Line SHAWN FERGUSON,* RICHARD L. HEBERT,*´ † and ODETTE LANEUVILLE‡ Departments of *Cellular and Molecular Medicine, †Medicine, and ‡Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, Ontario, Canada. Abstract. The cortical collecting duct (CCD) is a major site of Western blot analysis, COX-2 expression was significantly intrarenal prostaglandin E2 (PGE2) synthesis. This study ex- upregulated by incubation with either indomethacin or NS-398. amines the expression and regulation of the prostaglandin These drugs did not affect COX-1 protein expression. Evalu- synthesizing enzymes cyclooxygenase-1 (COX-1) and -2 in the ation of COX-2 mRNA expression by Northern blot analysis CCD. By indirect immunofluorescence using isoform-specific after NS-398 treatment demonstrated that the COX-2 protein antibodies, COX-1 and -2 immunoreactivity was localized to upregulation occurred independently of any change in COX-2 all cell types of the murine M-1 CCD cell line. By immuno- mRNA expression. These studies have for the first time local- histochemistry, both COX-1 and COX-2 were localized to ized COX-2 to the CCD and provided evidence that the inter- intercalated cells of the CCD on paraffin-embedded mouse calated cells of the CCD express both COX-1 and COX-2. The kidney sections. When COX enzyme activity was measured in results also demonstrate that constitutively expressed COX-2 is the M-1 cells, both indomethacin (COX-1 and -2 inhibitor) and the major COX isoform contributing to PGE2 synthesis by the the specific COX-2 inhibitor NS-398 effectively blocked PGE2 M-1 CCD cell line.