The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Homologous Isofunctional Enzymes: a Systematic Analysis Of
Omelchenko et al. Biology Direct 2010, 5:31 http://www.biology-direct.com/content/5/1/31 RESEARCH Open Access Non-homologousResearch isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution Marina V Omelchenko, Michael Y Galperin*, Yuri I Wolf and Eugene V Koonin Abstract Background: Evolutionarily unrelated proteins that catalyze the same biochemical reactions are often referred to as analogous - as opposed to homologous - enzymes. The existence of numerous alternative, non-homologous enzyme isoforms presents an interesting evolutionary problem; it also complicates genome-based reconstruction of the metabolic pathways in a variety of organisms. In 1998, a systematic search for analogous enzymes resulted in the identification of 105 Enzyme Commission (EC) numbers that included two or more proteins without detectable sequence similarity to each other, including 34 EC nodes where proteins were known (or predicted) to have distinct structural folds, indicating independent evolutionary origins. In the past 12 years, many putative non-homologous isofunctional enzymes were identified in newly sequenced genomes. In addition, efforts in structural genomics resulted in a vastly improved structural coverage of proteomes, providing for definitive assessment of (non)homologous relationships between proteins. Results: We report the results of a comprehensive search for non-homologous isofunctional enzymes (NISE) that yielded 185 EC nodes with two or more experimentally characterized - or predicted - structurally unrelated proteins. Of these NISE sets, only 74 were from the original 1998 list. Structural assignments of the NISE show over-representation of proteins with the TIM barrel fold and the nucleotide-binding Rossmann fold. From the functional perspective, the set of NISE is enriched in hydrolases, particularly carbohydrate hydrolases, and in enzymes involved in defense against oxidative stress. -

S41598-018-28214-2.Pdf
www.nature.com/scientificreports OPEN Dissecting Distinct Roles of NEDDylation E1 Ligase Heterodimer APPBP1 and UBA3 Received: 7 November 2017 Accepted: 25 May 2018 Reveals Potential Evolution Process Published: xx xx xxxx for Activation of Ubiquitin-related Pathways Harbani Kaur Malik-Chaudhry1, Zied Gaieb1, Amanda Saavedra1, Michael Reyes1, Raphael Kung1, Frank Le1, Dimitrios Morikis1,2 & Jiayu Liao1,2 Despite the similar enzyme cascade in the Ubiquitin and Ubiquitin-like peptide(Ubl) conjugation, the involvement of single or heterodimer E1 activating enzyme has been a mystery. Here, by using a quantitative Förster Resonance Energy Transfer (FRET) technology, aided with Analysis of Electrostatic Similarities Of Proteins (AESOP) computational framework, we elucidate in detail the functional properties of each subunit of the E1 heterodimer activating-enzyme for NEDD8, UBA3 and APPBP1. In contrast to SUMO activation, which requires both subunits of its E1 heterodimer AOS1-Uba2 for its activation, NEDD8 activation requires only one of two E1 subunits, UBA3. The other subunit, APPBP1, only contributes by accelerating the activation reaction rate. This discovery implies that APPBP1 functions mainly as a scafold protein to enhance molecular interactions and facilitate catalytic reaction. These fndings for the frst time reveal critical new mechanisms and a potential evolutionary pathway for Ubl activations. Furthermore, this quantitative FRET approach can be used for other general biochemical pathway analysis in a dynamic mode. Ubiquitin and Ubls are peptides that are conjugated to various target proteins to either lead the targeted protein to degradation or changes of activities in vivo, and their dysregulations ofen leads to various diseases, such as can- cers or neurodegenerative diseases1–3. -

Open Thesis Master Document V5.0.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Science IDENTIFICATION OF ENDOGENOUS MODULATORS FOR THE ARYL HYDROCARBON RECEPTOR A Thesis in Genetics by Christopher R. Chiaro © 2007 Christopher R. Chiaro Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December, 2007 The thesis of Christopher R. Chiaro was reviewed and approved* by the following: Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Thesis Advisor Chair of Committee C. Channa Reddy Distinguished Professor of Veterinary Science A. Daniel Jones Senior Scientist Department of Chemistry John P. Vanden Heuvel Professor of Veterinary Science Richard Ordway Associate Professor of Biology Chair of Genetics Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor capable of being regulated by a structurally diverse array of chemicals ranging from environmental carcinogens to dietary metabolites. A member of the basic helix-loop- helix/ Per-Arnt-Sim (bHLH-PAS) super-family of DNA binding regulatory proteins, the AhR is an important developmental regulator that can be detected in nearly all mammalian tissues. Prior to ligand activation, the AhR resides in the cytosol as part of an inactive oligomeric protein complex comprised of the AhR ligand-binding subunit, a dimer of the 90 kDa heat shock protein, and a single molecule each of the immunophilin like X-associated protein 2 (XAP2) and p23 proteins. Functioning as chemosensor, the AhR responds to both endobiotic and xenobiotic derived chemical ligands by ultimately directing the expression of metabolically important target genes. -

Biosynthesis in Vitro of Bacillamide Intermediate-Heterocyclic Alacysthiazole by Heterologous Expression of Nonribosomal Peptide Synthetase (NRPS) T
Journal of Biotechnology 292 (2019) 5–11 Contents lists available at ScienceDirect Journal of Biotechnology journal homepage: www.elsevier.com/locate/jbiotec Biosynthesis in vitro of bacillamide intermediate-heterocyclic AlaCysthiazole by heterologous expression of nonribosomal peptide synthetase (NRPS) T Fengli Zhang, Nayila Mulati, Yukun Wang, Yingxin Li, Sanqiang Gong, Loganathan Karthik, ⁎ Wei Sun, Zhiyong Li Marine Biotechnology Laboratory, State Key Laboratory of Microbial Metabolism and School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai, China ARTICLE INFO ABSTRACT Keywords: Bacillamide C, a potential natural antialgae active compound, is produced by Bacillus atrophaeus C89 derived Bacillus atrophaeus from marine sponge Dysidea avara. A nonribosomal peptide synthetase (NRPS) cluster is hypothesized to be Bacillamides involved in the biosynthesis of bacillamide C. The NRPS with a domain string of A1-PCP1-Cy-A2-PCP2-C can be Heterologous expression divided into three functional modules. After heterologous expression and purification of module A1-PCP1 and Nonribosomal peptide synthetase (NRPS) module Cy-A2-PCP2, their catalytic activities were biochemically proven in vitro by the reaction with the apo- Thiazole PCP domain transformed to the holo-PCP domain through a phosphopantetheinyl transferase, ATP, and substrate amino acids. Five– membered heterocyclic AlaCysthiazole with molecular weight of 172.0389 was detected. This proved the formation of the heterocyclic dipeptide AlaCysthiazole, which is considered to be a building block for the biosynthesis of bacillamide. This study provides a basis for further biosynthesis of bacillamides. 1. Introduction et al., 2017). Even though the biosynthesis of bacillamide C was opti- mized, the yield was very low (Jin et al., 2011; Yu et al., 2015). -

The Anti-Tumor Activity of the NEDD8 Inhibitor Pevonedistat in Neuroblastoma
International Journal of Molecular Sciences Article The Anti-Tumor Activity of the NEDD8 Inhibitor Pevonedistat in Neuroblastoma Jennifer H. Foster 1,* , Eveline Barbieri 1, Linna Zhang 1, Kathleen A. Scorsone 1, Myrthala Moreno-Smith 1, Peter Zage 2,3 and Terzah M. Horton 1,* 1 Texas Children’s Cancer and Hematology Centers, Department of Pediatrics, Section of Hematology and Oncology, Baylor College of Medicine, Houston, TX 77030, USA; [email protected] (E.B.); [email protected] (L.Z.); [email protected] (K.A.S.); [email protected] (M.M.-S.) 2 Department of Pediatrics, Division of Hematology-Oncology, University of California San Diego, La Jolla, CA 92024, USA; [email protected] 3 Peckham Center for Cancer and Blood Disorders, Rady Children’s Hospital, San Diego, CA 92123, USA * Correspondence: [email protected] (J.H.F.); [email protected] (T.M.H.); Tel.: +1-832-824-4646 (J.H.F.); +1-832-824-4269 (T.M.H.) Abstract: Pevonedistat is a neddylation inhibitor that blocks proteasomal degradation of cullin–RING ligase (CRL) proteins involved in the degradation of short-lived regulatory proteins, including those involved with cell-cycle regulation. We determined the sensitivity and mechanism of action of pevonedistat cytotoxicity in neuroblastoma. Pevonedistat cytotoxicity was assessed using cell viability assays and apoptosis. We examined mechanisms of action using flow cytometry, bromod- eoxyuridine (BrDU) and immunoblots. Orthotopic mouse xenografts of human neuroblastoma were generated to assess in vivo anti-tumor activity. Neuroblastoma cell lines were very sensitive to pevonedistat (IC50 136–400 nM). The mechanism of pevonedistat cytotoxicity depended on p53 Citation: Foster, J.H.; Barbieri, E.; status. -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -
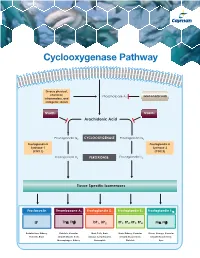
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Chlamydia Trachomatis-Containing Vacuole Serves As Deubiquitination
RESEARCH ARTICLE Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense Annette Fischer1, Kelly S Harrison2, Yesid Ramirez3, Daniela Auer1, Suvagata Roy Chowdhury1, Bhupesh K Prusty1, Florian Sauer3, Zoe Dimond2, Caroline Kisker3, P Scott Hefty2, Thomas Rudel1* 1Department of Microbiology, Biocenter, University of Wu¨ rzburg, Wu¨ rzburg, Germany; 2Department of Molecular Biosciences, University of Kansas, lawrence, United States; 3Rudolf Virchow Center for Experimental Biomedicine, University of Wu¨ rzburg, Wu¨ rzburg, Germany Abstract Obligate intracellular Chlamydia trachomatis replicate in a membrane-bound vacuole called inclusion, which serves as a signaling interface with the host cell. Here, we show that the chlamydial deubiquitinating enzyme (Cdu) 1 localizes in the inclusion membrane and faces the cytosol with the active deubiquitinating enzyme domain. The structure of this domain revealed high similarity to mammalian deubiquitinases with a unique a-helix close to the substrate-binding pocket. We identified the apoptosis regulator Mcl-1 as a target that interacts with Cdu1 and is stabilized by deubiquitination at the chlamydial inclusion. A chlamydial transposon insertion mutant in the Cdu1-encoding gene exhibited increased Mcl-1 and inclusion ubiquitination and reduced Mcl- 1 stabilization. Additionally, inactivation of Cdu1 led to increased sensitivity of C. trachomatis for IFNg and impaired infection in mice. Thus, the chlamydial inclusion serves as an enriched site for a *For correspondence: thomas. deubiquitinating activity exerting a function in selective stabilization of host proteins and [email protected]. protection from host defense. de DOI: 10.7554/eLife.21465.001 Competing interests: The authors declare that no competing interests exist. -

The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis
Article The COP9 Signalosome Variant CSNCSN7A Stabilizes the Deubiquitylating Enzyme CYLD Impeding Hepatic Steatosis Xiaohua Huang 1,* , Dawadschargal Dubiel 2 and Wolfgang Dubiel 2,3,*,† 1 Charité—Universitätsmedizin Berlin, Chirurgische Klinik, Campus Charité Mitte|Campus Virchow-Klinikum, Experimentelle Chirurgie und Regenerative Medizin, Augustenburger Platz 1, 13353 Berlin, Germany 2 Institute of Experimental Internal Medicine, Medical Faculty, Otto von Guericke University, Leipziger Str. 44, 39120 Magdeburg, Germany; [email protected] 3 State Key Laboratory of Cellular Stress Biology, Fujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, Xiang’an South Road, Xiamen 361102, China * Correspondence: [email protected] (X.H.); [email protected] (W.D.) † Lead Contact. Abstract: Hepatic steatosis is a consequence of distorted lipid storage and plays a vital role in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). This study aimed to explore the role of the COP9 signalosome (CSN) in the development of hepatic steatosis and its interplay with the deubiquitylating enzyme (DUB) cylindromatosis (CYLD). CSN occurs as CSNCSN7A and CSNCSN7B variants regulating the ubiquitin proteasome system. It is a deneddylating complex and associates with other DUBs. CYLD cleaves Lys63-ubiquitin chains, regulating a signal cascade that mitigates hepatic steatosis. CSN subunits CSN1 and CSN7B, as well as CYLD, were downregulated with specific siRNA in HepG2 cells and human primary hepatocytes. The same cells were transfected Citation: Huang, X.; Dubiel, D.; with Flag-CSN7A or Flag-CSN7B for pulldowns. Hepatic steatosis in cell culture was induced Dubiel, W. The COP9 Signalosome by palmitic acid (PA). Downregulation of CSN subunits led to reduced PPAR-γ expression. -

NEDD8 Acts As a 'Molecular Switch' Defining the Functional Selectivity Of
scientificscientificreport report NEDD8 acts as a ‘molecular switch’ defining the functional selectivity of VHL Ryan C. Russell & Michael Ohh+ Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, Canada The von Hippel–Lindau (VHL) tumour suppressor protein is (HIF), which is the main transcription factor that activates the important in the E3 ubiquitin ligase ECV (Elongin B/C–CUL2– expression of many hypoxia-inducible genes to counter VHL)-mediated destruction of hypoxia-inducible factor and the the detrimental effects of compromised oxygen availability promotion of fibronectin (FN) extracellular matrix assembly. (Kaelin, 2002), and as a positive regulator of fibronectin (FN) Although the precise molecular mechanism controlling the extracellular matrix (ECM) assembly (Roberts & Ohh, 2008). selectivity of VHL function remains unknown, a failure in either ECV (Elongin B/C–CUL2–VHL) is an SCF (SKP1/CDC53 or process is associated with oncogenic progression. Here, we show CUL1/F-box protein)-like E3 ubiquitin ligase in which VHL acts as that VHL performs its FN-associated function independently of a substrate-conferring component that recruits the a-subunits of the ECV complex, highlighting the autonomy of these pathways. HIF. These have been modified with hydroxyl groups on Furthermore, we show that NEDD8, a ubiquitin-like molecule, conserved prolyl residues within the oxygen-dependent degrada- acts as a ‘molecular switch’ in which its covalent conjugation to tion domain (ODD) by a class of prolyl hydroxylases in an oxygen- VHL prohibits the engagement of the scaffold component CUL2 dependent manner (Kaelin, 2002). This mechanistic insight and, concomitantly, activates the association with FN. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota.