University of Groningen Reflections on Flurbiprofen Eyedrops Van Sorge
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

Dentistry and Basic Non- Opioid Prescribing in Pain Dmitry M
Dentistry and Basic Non- Opioid Prescribing in Pain Dmitry M. Arbuck, MD President, Indiana Polyclinic Clinical Associate Professor of Psychiatry and Pain Management, Marian University College of Osteopathic Medicine Clinical Assistant Professor of Psychiatry and Medicine, IU School of Medicine www.IndianaPolyclinic.com Version May 2020 1 Disclosures No disclosures currently (May 7, 2020) 2 Disclaimer ISDH Oral Health Program Disclaimer for courses or presentations: The information provided in this course or presentation does not, and is not intended to, constitute dental, medical, or legal advice; instead, all information, content, and materials available in this course or presentation are for general informational purposes only. You should contact an outside dentist, physician, or attorney to obtain dental, medical, or legal advice and prior to acting, or refraining from acting, on the basis of information contained in this course or presentation. All liability with respect to actions taken or not taken based on the contents of this course or presentation are hereby expressly disclaimed. 3 Goals of Pain Management • Decrease pain • Increase function • Utilize medications that limit unacceptable side effects, including addiction 4 Goals of This Presentation • Gain knowledge of appropriate use of NSAIDs and acetaminophen for pain management in dentistry • Improve insight into benefits and adverse effects of various NSAIDs • Learn appropriate alternatives to opioid use for pain management 5 Opioids: Use with Caution • Use of opioids for -

Open Thesis Master Document V5.0.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Science IDENTIFICATION OF ENDOGENOUS MODULATORS FOR THE ARYL HYDROCARBON RECEPTOR A Thesis in Genetics by Christopher R. Chiaro © 2007 Christopher R. Chiaro Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December, 2007 The thesis of Christopher R. Chiaro was reviewed and approved* by the following: Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Thesis Advisor Chair of Committee C. Channa Reddy Distinguished Professor of Veterinary Science A. Daniel Jones Senior Scientist Department of Chemistry John P. Vanden Heuvel Professor of Veterinary Science Richard Ordway Associate Professor of Biology Chair of Genetics Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor capable of being regulated by a structurally diverse array of chemicals ranging from environmental carcinogens to dietary metabolites. A member of the basic helix-loop- helix/ Per-Arnt-Sim (bHLH-PAS) super-family of DNA binding regulatory proteins, the AhR is an important developmental regulator that can be detected in nearly all mammalian tissues. Prior to ligand activation, the AhR resides in the cytosol as part of an inactive oligomeric protein complex comprised of the AhR ligand-binding subunit, a dimer of the 90 kDa heat shock protein, and a single molecule each of the immunophilin like X-associated protein 2 (XAP2) and p23 proteins. Functioning as chemosensor, the AhR responds to both endobiotic and xenobiotic derived chemical ligands by ultimately directing the expression of metabolically important target genes. -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

SOME NEW DRUGS in the TREATMENT of RHEUMATIC FEVER by M
Postgrad Med J: first published as 10.1136/pgmj.28.317.179 on 1 March 1952. Downloaded from I79 SOME NEW DRUGS IN THE TREATMENT OF RHEUMATIC FEVER By M. J. H. SMITH, M.PHARM., PH.D., F.R.I.C. Department of Chemical Pathology, King's College Hospital Medical School, London Introduction every 4 to 8 hours. Symptoms such as dizziness, The usefulness of salicylates in rheumatic fever drowsiness and nausea developed in a small pro- is unquestioned, though their undesirable side- portion of the subjects, but in no instance were effects on the gastro-intestinal tract and on the these side-effects serious. The substance differed special senses are a drawback in prolonged therapy. from salicylic acid in producing a depression of Attempts to find allied substances with a greater the central nervous system in laboratory animals safety margin have been made and three com- and a decrease in the prothrombin time in man. pounds, salicylamide, sodium gentisate and The favourable clinical reports have led to the y-resorcylic acid, have recently been introduced. proposal that a large well-controlled trial should The treatment of rheumatic fever with ACTH be made.6 and cortisone has been the subject of a number of general reviews1' 2 and will not be discussed in the Gentisic Acid (2: 5-dihydroxybenzoic acid) present article. The cost and scarcity of these COOH by copyright. materials have stimulated a search for simpler compounds with a similar physiological action and a cinchoninic acid derivative (HPC) for which an ACTH-like activity is claimed, has been tried \AOH clinically in acute rheumatic fever. -

Colonoscopy Instructions
Colonoscopy Checklist Five days before your colonoscopy: Stop any medications that thin the blood (see list below) Discuss the discontinuation of these medications with your primary care physician to ensure that it is safe to stop them Three days before your colonoscopy: Stop eating high fiber foods including nuts, corn, popcorn, raw fruits, vegetables, and bran Stop fiber supplements The day before your colonoscopy: Have a normal breakfast If your colonoscopy is scheduled before noon the following day, do not have any lunch If your colonoscopy is scheduled after noon, have a light lunch Have clear liquids for the rest of the day (see below) Start prep as instructed by your physician Do not have anything to eat or drink after midnight The day of your colonoscopy: Take your blood pressure medications with a sip of water Make sure you bring your driver’s license or photo ID and leave valuables and jewelry at home Clear Liquid Diet Water Any kind of soft drink (ginger ale, cola, tonic, etc) Gatorade Apple Juice Orange Juice without pulp Lemonade Tea/Coffee (without milk) Dietary supplements (Ensure, Boost, Enlive, etc) Clear broth (vegetable, chicken, or beef) Jell‐O (stay away from red, blue, or purple colors) Ice pops without milk or fruit bits Honey or sugar NO DAIRY PRODUCTS Medications to stop prior to colonoscopy Below is a list of many medications (but not all) that fall into these categories. It is important to remember that there are hundreds of over‐the‐counter medications that contain NSAIDs or aspirin, so it is important to carefully read the label of any medication that you are taking (prescription or over‐the‐counter). -
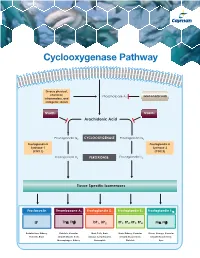
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Be Aware of Some Medications
Aspirin, Pain Relievers, Cold and Fever Remedies and Arthritis Medications when you have asthma, rhinitis or nasal polyps For some people with asthma, rhinitis and nasal polyps, medications such as acetylsalicylic acid or ASA and some arthritis medications can trigger very severe asthma, rhinitis, hives, swelling and shock. If you react to one of these medications, you will probably react to all of the others as well. There are many medications and products that contain ASA. This handout names some. Since new products are coming out all of the time, it is best to check with the pharmacist before using. Check the label yourself as well. If you react to these medications you must: Avoid these medications and products at all times. Let your doctor know right away if you have taken one of the medications and develop symptoms. Check the ingredients on the label yourself. Ask your pharmacist, doctor or other health care provider if you have questions. Get a medical alert bracelet or card that says you are allergic to ASA. Tell all of your health care providers that you are allergic to ASA. For pain control, use acetaminophen. Some products with acetaminophen are Atasol, Tempra, Tylenol and Novo-Gesic. Most people allergic to ASA can use acetaminophen. Firestone Institute for Respiratory Health St. Joseph’s Hospital McMaster University Health Sciences Some Products that Contain ASA 217s, 217s Strong D P 222s Doan’s Backache Pills Pepto-Bismol 282s, 282s Meps Dodd’s Tablets – All Percodan 292s Dolomine 37 Phenaphen with Codeine 692s Dristan Extra Strength PMS-Sulfasalazine A Dristan – All kinds AC&C R AC&C Extra Strength E Ratio-Oxycodan Acetylsalicylic Acid EC ASA Relief ASA Achrocidin Ecotrin – All kinds Robaxisal – All kinds Aggrenox Endodan Alka Seltzer – All kinds Enteric coated ASA S Anacin – All kinds Enteric coated aspirin Salazopyrin Antidol Entrophen – All kinds Salazopyrin Enema Apo-ASA Excedrin Salofalk Enema Apo-Asen Sulfasalazine Arco Pain Tablet F Arthrisin Fiorinal T Artria SR Fiorinal with Codeine Tri-Buffered ASA ASA, A.S.A. -

Of 20 PRODUCT MONOGRAPH FLURBIPROFEN Flurbiprofen Tablets BP 50 Mg and 100 Mg Anti-Inflammatory, Analgesic Agent AA PHARM
PRODUCT MONOGRAPH FLURBIPROFEN Flurbiprofen Tablets BP 50 mg and 100 mg Anti-inflammatory, analgesic agent AA PHARMA INC. DATE OF PREPARATION: 1165 Creditstone Road, Unit #1 April 16, 1991 Vaughan, Ontario L4K 4N7 DATE OF REVISION: February 7, 2019 Submission Control No. 223098 Page 1 of 20 PRODUCT MONOGRAPH NAME OF DRUG FLURBIPROFEN Flurbiprofen Tablets BP 50 mg and 100 mg PHARMACOLOGICAL CLASSIFICATION Anti-inflammatory, analgesic agent ACTIONS AND CLINICAL PHARMACOLOGY FLURBIPROFEN (flurbiprofen), a phenylalkanoic acid derivative, is a non-steroidal anti- inflammatory agent which also possesses analgesic and antipyretic activities. Its mode of action, like that of other non-steroidal anti-inflammatory agents, is not known. However, its therapeutic action is not due to pituitary adrenal stimulation. Flurbiprofen is an inhibitor of prostaglandin synthesis. The resulting decrease in prostaglandin synthesis may partially explain the drug's anti-inflammatory effect at the cellular level. Pharmacokinetics: Flurbiprofen is well absorbed after oral administration, reaching peak blood levels in approximately 1.5 hours (range 0.5 to 4 hours). Administration of flurbiprofen with food does not alter total drug availability but delays absorption. Excretion of flurbiprofen is virtually complete 24 hours after the last dose. The elimination half-life is 5.7 hours with 90% of the half-life values from 3-9 hours. There is no evidence of drug accumulation and flurbiprofen does not induce enzymes that alter its metabolism. Flurbiprofen is rapidly metabolized and excreted in the urine as free and unaltered intact drug (20-25%) and hydroxylated metabolites (60-80%). In animal models of inflammation the metabolites showed no activity. -

A COMPARATIVE STUDY of FLURBIPROFEN and ASPIRIN in SOFT TISSUE TRAUMA Accident Service, Radcliffe Infirmary, Oxford Surgeon
Br J Sports Med: first published as 10.1136/bjsm.10.1.11 on 1 March 1976. Downloaded from 11 A COMPARATIVE STUDY OF FLURBIPROFEN AND ASPIRIN IN SOFT TISSUE TRAUMA David S. MUCKLE, M.B., B.S., F.R.C.S., M.S. Accident Service, Radcliffe Infirmary, Oxford Surgeon and Medical Adviser to Oxford United F. C. ABSTRACT A double blind study using flurbiprofen (2-(2-fluoro-4-biphenylyl propionic acid) 150 mg daily and soluble aspirin (3.6 g daily) for 5 days immediately after injury, was carried out in 52 soft tissue injuries to the lower limb in professional sportsmen. Flurbiprofen was more effective than aspirin in producing analgesia (when daily pain scores were considered) after day 2 (p < 0.02); and flurbiprofen produced a more effective resolution of soft tissue trauma when days to training and match play were considered (p < 0.05). The inhibitory effects of flurbiprofen on prostaglandin biosynthesis and tissue action are mentioned and the use of anti-inflammatory agents given immediately after soft tissue injuries discussed. Introduction level of pain (mild, moderate or severe) according to instructions outlined on the jacket of the pack, and the In a previous double blind study using a phenylal- appropriate square was marked. The injured area was kanoic acid, "Brufen", (ibuprofen (2-(4-iso-butylphenyl) observed daily by the trainer or club doctor and all signs propionic acid) 1,200 mg daily) and soluble aspirin (3.0 recorded. The dates to full training and match fitness g daily) it was shown that the immediate use of these were noted. -

Topical Pharmaceutical Compositions of Flurbiprofen and Methyl Salicylate
(19) & (11) EP 2 455 074 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.05.2012 Bulletin 2012/21 A61K 31/05 (2006.01) A61K 47/10 (2006.01) A61K 9/00 (2006.01) (21) Application number: 11187973.0 (22) Date of filing: 04.11.2011 (84) Designated Contracting States: • Türkyilmaz, Ali AL AT BE BG CH CY CZ DE DK EE ES FI FR GB 34398 Istanbul (TR) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • Akalin, Nur Pehlivan PL PT RO RS SE SI SK SM TR 34398 Istanbul (TR) Designated Extension States: • Önder, Ramazan BA ME 34398 Istanbul (TR) • Öner, Levent (30) Priority: 08.11.2010 TR 201009220 Ankara (TR) (71) Applicant: Sanovel Ilac Sanayi ve Ticaret A.S. (74) Representative: Sevinç, Erkan 34398 Istanbul (TR) Istanbul Patent & Trademark Consultancy Ltd. Plaza-33, Büyükdere cad. No: 33/16, Sisli (72) Inventors: 34381 Istanbul (TR) • Cifter, Ümit 34398 Istanbul (TR) (54) Topical pharmaceutical compositions of flurbiprofen and methyl salicylate (57) The present invention relates to topical pharma- thermore, the invention relates to process for preparing ceutical compositions comprising flurbiprofen or a phar- the said topical pharmaceutical compositions and its use maceutically acceptable salt thereof and methyl sali- for the treatment of pain and inflammatory symptoms as- cylate. More specifically, the invention relates to topical sociated with muscle-skeletol system, joint and soft-tis- pharmaceutical compositions of flurbiprofen and methyl sue disorders. salicylate characterized in that said composition compris- es dimethyl sulfoxide and one or more gelling agent.