Prostacyclin: an Inflammatory Paradox
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

AA Metabolism, 139 A2b1, 69, 70, 318–323 ABCB1 Gene, 176 Abciximab
Index A aIIbb3, 60, 62, 69, 70, 73, 74, 79, 80 AA. See Arachidonic acid (AA) AJW202, 293 AA metabolism, 139 Akt-1, 345 a2b1, 69, 70, 318–323 Akt-2, 345 ABCB1 gene, 176 ALX-0081, 296 Abciximab, 206–208, 497, 500, 501, 504–505, ALX-0681, 298 509, 513, 514, 525 Anagrelide, 227, 229 approval, 206 Ankle brachial indexes (ABIs), 549, 587, EPIC trial, 207 595–597 EPILOG study, 207 framingham risk score, 551 EPISTENT study, 207 screening, 550 platelet binding, 207 sensitivity, 551 thrombocytopenia, 206 specificity, 551 in unstable angina, 208 vascular risk, 550 ABIs. See Ankle brachial indexes (ABIs) Antagomirs, 439 Acetylsalicylic acid (aspirin), 137–158 Antibodies, 317 ACS. See Acute coronary syndromes (ACS) Anticoagulants, 525, 537 Acute coronary syndromes (ACS), 288 Anticoagulation, 533, 534 Acute ischemic stroke, 143, 149–150 Antioxidant, 233 Acute myocardial infarction, 142, 143, 149, Antiplatelet agents, 263, 553 153, 157 Antithrombotic Trialist’s ADAMTS-13, 93 Collaboration, 553 Adenosine diphosphate (ADP), 37, 88, 96, aspirin, 553 166, 473 clopidogrel, 553 Adenosine triphosphate (ATP), 37 dipyridamole, 553 Adhesion, 90, 112–118, 125 meta-analysis, 560 Adhesion molecules infiltration, 264 picotamide, 553 Adiponectin, 315 risk reduction, 553 ADP. See Adenosine diphosphate (ADP) ticlopidine, 553 ADP inhibitors (or P2Y12 blockers), 497–499, vascular events, 553 501–505, 507–509, 513, 514 Antiplatelet therapy, 472 ADP-receptor antagonists, 169 Apixaban, 535, 539 Adverse effects, 153–156 Aptamers, 299–301 Aegyptin, 327 Arachidonic acid (AA), 474 P. Gresele et al. (eds.), Antiplatelet Agents, Handbook of Experimental 607 Pharmacology 210, DOI 10.1007/978-3-642-29423-5, # Springer-Verlag Berlin Heidelberg 2012 608 Index ARC1779, 299–300 TXS signaling, 279 ARC15105, 300 Cardiovascular death, 248 AR-C69931MX, 456 Carotid endarterectomy (ACE) inhibition, Aspirin, 474, 496, 498–499, 501–502, 143, 156 504–507, 511–514, 520, 522, Carotid stenosis, 523 527, 531, 533–537, 539, CCBs. -

Open Thesis Master Document V5.0.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Science IDENTIFICATION OF ENDOGENOUS MODULATORS FOR THE ARYL HYDROCARBON RECEPTOR A Thesis in Genetics by Christopher R. Chiaro © 2007 Christopher R. Chiaro Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December, 2007 The thesis of Christopher R. Chiaro was reviewed and approved* by the following: Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Thesis Advisor Chair of Committee C. Channa Reddy Distinguished Professor of Veterinary Science A. Daniel Jones Senior Scientist Department of Chemistry John P. Vanden Heuvel Professor of Veterinary Science Richard Ordway Associate Professor of Biology Chair of Genetics Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor capable of being regulated by a structurally diverse array of chemicals ranging from environmental carcinogens to dietary metabolites. A member of the basic helix-loop- helix/ Per-Arnt-Sim (bHLH-PAS) super-family of DNA binding regulatory proteins, the AhR is an important developmental regulator that can be detected in nearly all mammalian tissues. Prior to ligand activation, the AhR resides in the cytosol as part of an inactive oligomeric protein complex comprised of the AhR ligand-binding subunit, a dimer of the 90 kDa heat shock protein, and a single molecule each of the immunophilin like X-associated protein 2 (XAP2) and p23 proteins. Functioning as chemosensor, the AhR responds to both endobiotic and xenobiotic derived chemical ligands by ultimately directing the expression of metabolically important target genes. -

Estimation of Serum Prostaglandin D2 Levels and Its Expression in Tissue of Alopecia Areata
ISSN: 2536-9474 (Print) Original article / FYMJ ISSN: 2536-9482 (Online) Fayoum University Medical Journal Soliman et al., 2019,4(1), 77-85 Estimation of serum prostaglandin D2 levels and its expression in tissue of Alopecia areata Talal A. Abd-ElRaheem1, Samar M.R. El-Tahlawy2, Olfat G. Shaker3, Mohamed H.Mohamed4 and Yasmin F.Soliman5 1. M.D, professor of Dermatology, STDs and Andrology department, Faculty of Medicine Fayoum University. 2. M.D, professor of Dermatology department, Faculty of Medicine Cairo University 3. M.D, professor of Biochemistry department, Faculty of Medicine-Cairo University 4. MD, lecturer of Dermatology, STDs and Andrology department, Faculty of Medicine Fayoum University 5. M.B.B.CH, Dermatology, STDs and Andrology department, Faculty of Medicine, Cairo University Abstract Results: There was statistically highly Back ground: Alopecia areata is a recurrent, significant difference between the two groups non-scaring type of hair loss considered to be regarding the mean value of PGD2 in tissue in an autoimmune process. Though its AA patients. It was significantly lower than in etiopathogenesis is not fully understood, many control group (p < 0.001). The mean value of therapeutic options have been used by PGD2 in serum in AA patients was dermatologists, but none are curative or significantly lower than in control group (p< preventive. Prostaglandins analogues which 0.05). are used to treat glaucoma. Increase in eye lash Conclusion: Prostaglandin D2 exhibits a number, thickness and pigmentation have been strong role in etiology of alopecia areata and reported as side effect. significantly was elevated in serum and tissue Methods: This cross sectional case control of alopecia areata patients. -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

Effects of Selexipag and Its Active Metabolite in Contrasting The
Cutolo et al. Arthritis Research & Therapy (2018) 20:77 https://doi.org/10.1186/s13075-018-1577-0 RESEARCH ARTICLE Open Access Effects of selexipag and its active metabolite in contrasting the profibrotic myofibroblast activity in cultured scleroderma skin fibroblasts Maurizio Cutolo1*, Barbara Ruaro1, Paola Montagna1, Renata Brizzolara1, Emanuela Stratta2, Amelia Chiara Trombetta1, Stefano Scabini2, Pier Paolo Tavilla3, Aurora Parodi3, Claudio Corallo4, Nicola Giordano4, Sabrina Paolino1, Carmen Pizzorni1, Alberto Sulli1, Vanessa Smith5 and Stefano Soldano1 Abstract Background: Myofibroblasts contribute to fibrosis through the overproduction of extracellular matrix (ECM) proteins, primarily type I collagen (COL-1) and fibronectin (FN), a process which is mediated in systemic sclerosis (SSc) by the activation of fibrogenic intracellular signaling transduction molecules, including extracellular signal-regulated kinases 1 and 2 (Erk1/2) and protein kinase B (Akt). Selexipag is a prostacyclin receptor agonist synthesized for the treatment of pulmonary arterial hypertension. The study investigated the possibility for selexipag and its active metabolite (ACT-333679) to downregulate the profibrotic activity in primary cultures of SSc fibroblasts/myofibroblasts and the fibrogenic signaling molecules involved. Methods: Fibroblasts from skin biopsies obtained with Ethics Committee (EC) approval from patients with SSc, after giving signed informed consent, were cultured until the 3rd culture passage and then either maintained in normal growth medium (untreated cells) or independently treated with different concentrations of selexipag (from 30 μMto 0.3 μM) or ACT-333679 (from 10 μMto0.1μM) for 48 h. Protein and gene expressions of α-smooth muscle actin (α-SMA), fibroblast specific protein-1 (S100A4), COL-1, and FN were investigated by western blotting and quantitative real-time PCR. -
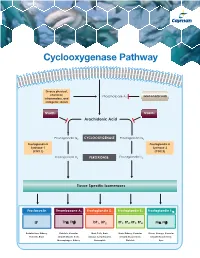
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

New Investigational Drugs for Androgenetic Alopecia. Valente Duarte De Sousa IC 1, Tosti A
Expert Opin Investig Drugs. 2013 May;22(5):573-89. doi: 10.1517/13543784.2013.784743. Epub 2013 Apr 4. New investigational drugs for androgenetic alopecia. Valente Duarte de Sousa IC 1, Tosti A . Author information • [email protected] Erratum in • Erratum. [Expert Opin Investig Drugs. 2015] Abstract INTRODUCTION: Androgenetic alopecia (AGA) is the most common form of hair loss, however current treatment options are limited and moderately effective. In the past few years, there has been an increased interest in deciphering the molecular mechanisms responsible for this disorder, which has opened the possibility of novel treatments that promise to not only stimulate hair growth, but also to induce formation of new hair follicles. AREAS COVERED: The future holds more effective topical treatments with less systemic side effects (such as topical 5- alfa-reductase inhibitors), prostaglandin analogs and antagonists, medications which act through the Wnt signaling pathway, stem cells for hair regeneration, platelet-rich plasma (PRP) and more effective ways of transplanting hair. A comprehensive search was made using PubMed, GoogleScholar and Clinicaltrial.gov using different combination of key words, which included AGA treatment, new treatments for AGA, Wnt pathway, prostaglandins, PRP and stem cells for hair regrowth. EXPERT OPINION: In the near future, treatments with topical 5-alfa-reductase inhibitors and prostaglandin agonists or antagonists are expected. More evidence is needed to verify the efficacy of PRP. Although hair follicle bioengineering and multiplication is a fascinating and promising field, it is still a long way from being available to clinicians. J Am Acad Dermatol. 2015 Apr;72(4):712-6. -

Effect of Prostanoids on Human Platelet Function: an Overview
International Journal of Molecular Sciences Review Effect of Prostanoids on Human Platelet Function: An Overview Steffen Braune, Jan-Heiner Küpper and Friedrich Jung * Institute of Biotechnology, Molecular Cell Biology, Brandenburg University of Technology, 01968 Senftenberg, Germany; steff[email protected] (S.B.); [email protected] (J.-H.K.) * Correspondence: [email protected] Received: 23 October 2020; Accepted: 23 November 2020; Published: 27 November 2020 Abstract: Prostanoids are bioactive lipid mediators and take part in many physiological and pathophysiological processes in practically every organ, tissue and cell, including the vascular, renal, gastrointestinal and reproductive systems. In this review, we focus on their influence on platelets, which are key elements in thrombosis and hemostasis. The function of platelets is influenced by mediators in the blood and the vascular wall. Activated platelets aggregate and release bioactive substances, thereby activating further neighbored platelets, which finally can lead to the formation of thrombi. Prostanoids regulate the function of blood platelets by both activating or inhibiting and so are involved in hemostasis. Each prostanoid has a unique activity profile and, thus, a specific profile of action. This article reviews the effects of the following prostanoids: prostaglandin-D2 (PGD2), prostaglandin-E1, -E2 and E3 (PGE1, PGE2, PGE3), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane-A2 (TXA2) on platelet activation and aggregation via their respective receptors. Keywords: prostacyclin; thromboxane; prostaglandin; platelets 1. Introduction Hemostasis is a complex process that requires the interplay of multiple physiological pathways. Cellular and molecular mechanisms interact to stop bleedings of injured blood vessels or to seal denuded sub-endothelium with localized clot formation (Figure1). -

Nanosuspensions of Selexipag: Formulation, Characterization, and in Vitro Evaluation Rusul M
Iraqi J Pharm Sci, Vol.30(1) 2021 Selexipag nanosuspension DOI : https://doi.org/10.31351/vol30iss1pp144-153 Nanosuspensions of Selexipag: Formulation, Characterization, and in vitro Evaluation Rusul M. Alwan*,1 and Nawal A. Rajab** * Ministry of Health and Environment, Najaf Health Department, Najaf, Iraq. **Department of Pharmaceutics, College of Pharmacy, University of Baghdad, Baghdad, Iraq. Abstract Selexipag(SLP) is an orally selective long-acting prostacyclin receptor agonist indicated for pulmonary arterial hypertension treatment. It is practically insoluble in water (class II, according to BCS). This work aims to prepare and optimize Selexipag nanosuspensions (SLPNS) to enhance the saturation solubility and in vitro dissolution rate. The solvent antisolvent precipitation method was used for the production of NS, and the effect of formulation parameters (stabilizer type, drug: stabilizer ratio, and use of co-stabilizer) and process parameter (stirring speed) on the particle size (P.S) and polydispersity index (PDI) were studied. The result revealed that the P.S of all prepared SLPNS formulation was in the nanometer range, except for the formulas that stabilized by Poloxamer. The optimal SLPNS (F15), which is stabilized by Soluplus® (SLP: stabilizer ratio 1:2) and prepared at a stirring speed of 1000 rpm, showed the smallest P.S and appropriate PDI, which are 47 nm and 0.073. The formula F5 exhibits 136 folds, an increase in the saturation solubility, and an enhancement in the dissolution rate in phosphate buffer pH 6.8 (100% drug release during 60 min) compared to the pure drug. This result indicates that SLPNS is an efficient way for improving the saturation solubility and the dissolution rate of SLP. -

Prostaglandin D2, a Neuromodulator
Proc. Natl. Acad. Sci. USA Vol. 76, No. 12, pp. 6231-6234, December 1979 Biochemistry Prostaglandin D2, a neuromodulator (prostaglandin D synthetase/enzyme distribution/neuroblastoma cell/cyclic AMP) TAKAO SHIMIZU, NOBORU MIZUNO*, TAKEHIKO AMANOt, AND OSAMU HAYAISHI Department of Medical Chemistry, and *Department of Anatomy, Kyoto University Faculty of Medicine, Sakyo-ku, Kyoto 606, Japan; and tMitsubishi-Kasei Institute of Life Sciences, Machida-shi, Tokyo 194, Japan Contributed by Osamu Hayaishi, September 17, 1979 ABSTRACT The distribution of prostaglandin D synthetase were quickly removed. The brain was chilled on ice and sepa- activity was determined in various tissues of rat by using the rated into 11 parts-cerebral neocortex, cerebellum, pons and supernatant fraction (10,000 X g, 20 min) of the homogenates. medulla oblongata, midbrain, hypothalamus, thalamus, bulbus The highest activity was found in brain, spinal cord, and ali- and mentary tract. The activity was ubiquitously distributed in all olfactorius, hippocampus, caudoputamen, pineal body, parts of brain, and the highest specific activity was found in meninges. These tissues were weighed and homogenized with hypothalamus and thalamus. Homogenates of two neuroblas- 2 vol of 10 mM potassium phosphate buffer (pH 6.0) containing toma cell lines were found to produce prostaglandin D2, 0.5 mM dithiothreitol in a Polytron homogenizer. Mouse neu- whereas a glioma cell line was almost inactive. Prostaglandin roblastoma cells (NS-20 and N1E-115) and rat glioma cells (C6 D2 is a potent and specific activator of the adenylate cyclase BU-1) (see below) were sonicated with a Branson Sonifier model system of cultured neuroblastoma cells, suggesting the possi- were bility that it may act as a neuromodulator in the central nervous W 185D (output 4, for 1.5 min).