Low-Copy Piggybac Transposon Mutagenesis in Mice Identifies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Epigenetic Loss of the RNA Decapping Enzyme NUDT16 Mediates C-MYC Activation in T-Cell Acute Lymphoblastic Leukemia
OPEN Leukemia (2017) 31, 1622–1657 www.nature.com/leu LETTERS TO THE EDITOR Epigenetic loss of the RNA decapping enzyme NUDT16 mediates C-MYC activation in T-cell acute lymphoblastic leukemia Leukemia (2017) 31, 1622–1625; doi:10.1038/leu.2017.99 Having found the aforementioned NUDT16 CpG island methy- lation profiles, we studied in greater detail their association with the possible transcriptional inactivation of the NUDT16 gene at the RNA and protein levels in leukemia cell lines. We first It is possible that the occurrence of intrinsic defects in RNA performed bisulfite genomic sequencing of mutiple clones in the – processing pathways, such as RNA decapping,1 3 contribute to the T-cell Acute Lymphoblastic Leukemia (T-ALL) cell lines CCRF-CEM, distorted RNA landscapes of cancer cells. After transcription by Jurkat, MOLT-4 and MOLT-16 using primers that encompassed the RNA polymerase II, RNA molecules are equipped with a 5´-end transcription start site-associated CpG island and confirmed the N7-methyl guanosine (m7G)-cap. This m7G-cap is essential for hypermethylated status of the 5′-end region of NUDT16 in translation, stabilizing the RNA molecule and protecting it from comparison to normal T lymphocytes (Figure 1b), validating the – exonucleolytic breakdown.1 3 For RNA decay to occur the DNA methylation patterns obtained by the microarray approach m7G-cap first needs to be removed. This process is known as (Supplementary Figure S2). In contrast, normal T lymphocytes, the – decapping.1 3 The decapping mRNA 2 (DCP2) enzyme,4 also T-ALL cell lines KOPN-8, REH and RS4;11 and the leukemia cell known as the nucleoside diphosphate-linked moiety X motif lines HL-60 and K562 derived from myeloid lineage were all found 20 (NUDT20), was originally thought to be the only mammalian to be unmethylated (Figure 1b; Supplementary Figure S2). -
C a B D G E F
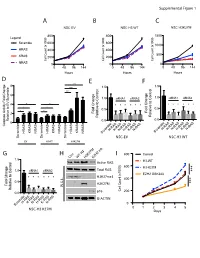
Supplemental Figure 1 A B C NSC EV NSC H3 WT NSC H3K27M 800 800 1500 ) ) Legend ) Scramble 600 600 x1000 x1000 x1000 ( ( ( 1000 t t t n n HRAS n 400 400 u u u o o o C C C 500 l l KRAS l 200 200 el el el C C C NRAS 0 0 0 0 48 96 144 0 48 96 144 0 48 96 144 Hours Hours Hours D *** E F 8 1.5 1.5 e *** l l *** ro ro t 6 t 1.0 siRNA1 siRNA2 1.0 siRNA1 siRNA2 Con Con o o t 4 * * t * * * * * * e * * * * * * e v i * * v i t 0.5 * * t 0.5 Fold Change 2 Fold Change Rela Rela Relative to EV Scrambl 0.0 0.0 0 Caspase Activity Fold Change H-RASK-RASN-RASH-RASK-RASN-RAS H-RASK-RASN-RASH-RASK-RASN-RAS KRAS KRAS KRAS HRAS NRAS HRAS NRAS HRAS NRAS Scramble Scramble Scramble Scramble Scramble NSC-EV NSC-H3 WT EV H3WT H3K27M G H I 800 Control Con WT H3 H3K27MEZH2 inb 1.5 l H3-WT Active RAS ro ) t 600 H3-K27M 1.0 siRNA1 siRNA2 Total RAS *** Con x1000 ( o EZH2 GSK343 t t * * * * * * H3K27me3 n e 400 u v i o t 0.5 *** H3K27M C Fold Change l W.C.L. el Rela p16 C 200 0.0 B-ACTIN H-RASK-RASN-RASH-RASK-RASN-RAS 0 Scramble NSC-H3 K27M 0 1 2 3 4 5 Days E C A 100 Cell Viability 100 25 50 75 Cell Viability 25 50 75 0 0 Fold Change MOCK MOCK Relative to Control Scramble * 0.0 0.2 0.4 0.6 0.8 1.0 1.2 MYC Scramble MYC MYC * PDGFRA Scramble siRNA PDGFRA MYC PDGFRA AURKA AURKA DIPG007siRNA2 PDGFRA NSC H3K27MsiRNA1and AURKA LAMTOR3 AURKA LAMTOR3 LAMTOR3 LIN28A LAMTOR3 LIN28A LIN28B LIN28A NSC-EV LIN28A MAP2K3 LIN28B LIN28B LIN28B MAP2K5 siRNA1 MAP2K3 MAP2K3 MAP3K2 MAP2K3 MAP3K7 MAP2K5 MAP2K5 MAPK7 MAP2K5 2 MAP3K2 siRNA1 siRNA2 MAP3K2 ZAK MAP3K7 MAP3K2 MAP3K7 MAPK7 MAP3K7 -

Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in Vitro and in Vivo
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Genetik Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in vitro and in vivo - Towards Response Prediction in Cancer Therapy with Kinase Inhibitors Michaela Bairlein Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ. -Prof. Dr. K. Schneitz Prüfer der Dissertation: 1. Univ.-Prof. Dr. A. Gierl 2. Hon.-Prof. Dr. h.c. A. Ullrich (Eberhard-Karls-Universität Tübingen) 3. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 07.01.2010 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 19.04.2010 angenommen. FOR MY PARENTS 1 Contents 2 Summary ................................................................................................................................................................... 5 3 Zusammenfassung .................................................................................................................................................... 6 4 Introduction .............................................................................................................................................................. 8 4.1 Cancer .............................................................................................................................................................. -

Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S
crossmark Research Author’s Choice © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S Rolf Turk‡§¶ʈ**, Jordy J. Hsiao¶, Melinda M. Smits¶, Brandon H. Ng¶, Tyler C. Pospisil‡§¶ʈ**, Kayla S. Jones‡§¶ʈ**, Kevin P. Campbell‡§¶ʈ**, and Michael E. Wright¶‡‡ Mutations in genes encoding components of the sar- The muscular dystrophies are hereditary diseases charac- colemmal dystrophin-glycoprotein complex (DGC) are re- terized primarily by the progressive degeneration and weak- sponsible for a large number of muscular dystrophies. As ness of skeletal muscle. Most are caused by deficiencies in such, molecular dissection of the DGC is expected to both proteins associated with the cell membrane (i.e. the sarco- reveal pathological mechanisms, and provides a biologi- lemma in skeletal muscle), and typical features include insta- cal framework for validating new DGC components. Es- bility of the sarcolemma and consequent death of the myofi- tablishment of the molecular composition of plasma- ber (1). membrane protein complexes has been hampered by a One class of muscular dystrophies is caused by mutations lack of suitable biochemical approaches. Here we present in genes that encode components of the sarcolemmal dys- an analytical workflow based upon the principles of pro- tein correlation profiling that has enabled us to model the trophin-glycoprotein complex (DGC). In differentiated skeletal molecular composition of the DGC in mouse skeletal mus- muscle, this structure links the extracellular matrix to the cle. We also report our analysis of protein complexes in intracellular cytoskeleton. -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

Targeted MS Quantitation Assays for Signal Transduction Protein Pathways
Targeted MS quantitation assays for signal transduction protein pathways Paul Haney R&D Platform Manager Thermo Scientific Protein Research Products Rockford, IL Why Targeted Quantitative Proteomics via Mass Spec? . Measure protein abundance and protein isoforms (e.g. splice variants, PTMs) without the need for antibodies . Avoid immuno-based cross- reactivity during multiplexing. Validate relative quantitation data from discovery proteomic experiments 2 How Do You Select Peptides for Targeted MS Assays? List of Proteins Spectral library Discovery data repositories In silico prediction Pinpoint 1.2 Hypothesis Experimental List of target peptides and transitions 3 How Do You Select Peptides for Targeted MS Assays? List of Proteins Spectral library Discovery data repositories In silico prediction Pinpoint 1.2 Hypothesis Experimental Validation Assay results List of target peptides and transitions 4 Tools for Target Peptide Identification and Scheduling . Active-site probes for enzyme subclass enrichment . Rapid recombinant heavy protein expression using human cell-free extracts . Peptide retention time calibration mixture for chromatography QC and targeted method 100 80 60 acquisition scheduling Intensity 40 20 0 5 10 15 20 25 30 5 Tools for Target Peptide Identification and Scheduling . Active-site probes for enzyme subclass enrichment . Rapid recombinant heavy protein expression using human cell-free extracts • Stergachis, A. & MacCoss, M. (2011) Nature Methods (submitted) . Peptide retention time calibration mixture for 100 80 chromatography -

Dema and Faust Et Al., Suppl. Material 2020.02.03
Supplementary Materials Cyclin-dependent kinase 18 controls trafficking of aquaporin-2 and its abundance through ubiquitin ligase STUB1, which functions as an AKAP Dema Alessandro1,2¶, Dörte Faust1¶, Katina Lazarow3, Marc Wippich3, Martin Neuenschwander3, Kerstin Zühlke1, Andrea Geelhaar1, Tamara Pallien1, Eileen Hallscheidt1, Jenny Eichhorst3, Burkhard Wiesner3, Hana Černecká1, Oliver Popp1, Philipp Mertins1, Gunnar Dittmar1, Jens Peter von Kries3, Enno Klussmann1,4* ¶These authors contributed equally to this work 1Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Robert- Rössle-Strasse 10, 13125 Berlin, Germany 2current address: University of California, San Francisco, 513 Parnassus Avenue, CA 94122 USA 3Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, 13125 Berlin, Germany 4DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Oudenarder Strasse 16, 13347 Berlin, Germany *Corresponding author Enno Klussmann Max Delbrück Center for Molecular Medicine Berlin in the Helmholtz Association (MDC) Robert-Rössle-Str. 10, 13125 Berlin Germany Tel. +49-30-9406 2596 FAX +49-30-9406 2593 E-mail: [email protected] 1 Content 1. CELL-BASED SCREENING BY AUTOMATED IMMUNOFLUORESCENCE MICROSCOPY 3 1.1 Screening plates 3 1.2 Image analysis using CellProfiler 17 1.4 Identification of siRNA affecting cell viability 18 1.7 Hits 18 2. SUPPLEMENTARY TABLE S4, FIGURES S2-S4 20 2 1. Cell-based screening by automated immunofluorescence microscopy 1.1 Screening plates Table S1. Genes targeted with the Mouse Protein Kinases siRNA sub-library. Genes are sorted by plate and well. Accessions refer to National Center for Biotechnology Information (NCBI, BLA) entries. The siRNAs were arranged on three 384-well microtitre platres. -

Downloaded from Phosphositeplus
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.22.215897; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. July 21, 2020 Phosphoproteomic Identification of Vasopressin-Regulated Protein Kinases in Collecting Duct Cells Arnab Datta1,2, Chin-Rang Yang1, Karim Salhadar1, Chung-Lin Chou1, Viswanathan Raghuram1, and Mark A. Knepper1 1Epithelial Systems Biology Laboratory, Systems Biology Center, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland 2Yenepoya Research Center, Yenepoya (Deemed to be University), University Road, Deralakatte, Mangalore 575018, Karnataka, India Running title: Vasopressin-regulated Kinases in Renal Collecting Duct Correspondence to Mark Knepper, MD, PhD, Division of Intramural Research, National Heart, Lung and Blood Institute, NIH, Bethesda, MD 20892 Phone: 301-496-3064; Email: [email protected] Keywords: mpkCCD, GPCR signaling, V2 receptor signaling, Desmopressin bioRxiv preprint doi: https://doi.org/10.1101/2020.07.22.215897; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT Background and Purpose: The peptide hormone vasopressin regulates water transport in the renal collecting duct largely via the V2 receptor, which triggers a cAMP-mediated activation of a protein kinase A (PKA)-dependent signaling network. -

Technische Universität München
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Proteomik und Bioanalytik Application of mass spectrometry-based proteomics to study cancer drug resistance mechanisms Heiner Matthias Koch Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Prof. Dr. D. Langosch Prüfer der Dissertation: 1. Prof. Dr. B. Küster 2. Prof. Dr. F. Bassermann Die Dissertation wurde am 12.07.2016 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 20.09.2016 angenommen. II Table of Content Abstract V Zusammenfassung VI Chapter I General Introduction 1 Chapter II Chemical proteomics uncovers EPHA2 as a mechanism 49 of acquired resistance to small molecule EGFR kinase inhibition Chapter III Phosphoproteome profiling reveals molecular mechanisms of 73 growth factor mediated kinase inhibitor resistance in EGFR overexpressing cancer cells Chapter IV Time resolved proteomic and phosphoproteomic analysis of 109 adaptation to kinase inhibition Chapter V General discussion 135 List of publications 144 Danksagung | Acknowledgment 145 Curriculum vitae 146 III IV Abstract In recent years an increasing number of small molecule kinase inhibitors were approved for targeted cancer therapies. Targeted therapies have less toxic side effects than conventional chemotherapeutics and promise efficacious personalized treatments. Although some molecules improved the outcome of selected patient groups, resistance almost invariably develops and represents a major clinical challenge. There is an intensive effort to circumvent emerging resistance by the development of new targeted agents or the combination of approved molecules. However, the molecular alterations that render cancer cells resistant are still poorly understood.