2. Chemistry and Physics of Transuranium Elements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

WO 2013/089962 Al 20 June 2013 (20.06.2013) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2013/089962 Al 20 June 2013 (20.06.2013) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every B01J 31/04 (2006.01) B01J 31/18 (2006.01) kind of national protection available): AE, AG, AL, AM, B01J 31/14 (2006.01) B01J 31/22 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (21) Number: International Application DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US20 12/065285 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 15 November 2012 (15.1 1.2012) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, (25) Filing Language: English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, (26) Publication Language: English TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 13/323,328 12 December 201 1 (12. 12.201 1) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): CHEV¬ GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, RON PHILLIPS CHEMICAL COMPANY LP UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, [US/US]; 10001 Six Pines Drive, The Woodlands, Texas TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, 77380 (US). -

AP Chemistry Summer Assignment 2021-22 School Year
AP Chemistry AP Chemistry Summer Assignment 2021-22 School Year Chemical Foundations Chapter 1 1. How many significant figures are in the following numbers? a. 0.00150 b. 0.1205 c. 200 d. 2.00 X 10 3 2. Complete the operation and report using the correct number of significant figures a. 26.20 - 0.5= b. 2.5 + 3.25 = c. .040 X 2.0 = d. 3.25 / 4 = e. 3.0 X 10 -3 X 6 = Atomic , Molecules Ions CH 2 and Stoichiometry CH 3 with Periodicity CH 7 3. What is an isotope? Refer to the isotope of Uranium 92U 4. How many protons and neutrons are in the nucleus of this isotope. 5. How many electrons are in a single atom of Uranium 6. What is the mass of this isotope of Uranium. 7. Assume silicon has three major isotopes in nature. The average atomic mass of silicon is 28.09 amu. Fill in the missing information in the table. Isotope Mass (amu) Abundance 28Si 27.89 29Si 4.70% 30Si 29.97 3.09% 8. Which color of light has the highest frequency, red or green ? 9. Which color of light has the longest wavelength, green or violet? 10. Hydrogen emits light with a wavelength of 410 nm, what is the frequency of this light? 11. What is the electron configuration, orbital notation and noble gas notation for phosphorus? For bromine? How many unpaired electrons does phosphorus have? How many unpaired electrons does bromine have? 12. What is the charge for a phosphorus ion? Why does it make this charge? 13. -

Chemical Hygiene Plan Ii Revised 03/2021 Table of Contents
MAR 2021 Office of Environmental Health and Safety Principal Author/Editor: David Webber, PhD/Chemical Hygiene Officer Contributing Authors/Editors: Nikolai Evdokimov, PhD, James Gibson, PhD, Tania Guardado, PhD, Amanda Jevons, Deona Willes, MPH Graphics/Design: Alfred M. Bouziane, MS, Brent Pantell USC Chemical Hygiene Plan ii Revised 03/2021 Table of Contents i.0 2021 Revision Summary Section 3.0 vi Section 4.0 vi Section 5.0 vii Section 7.0 vii Section 8.0 viii Section 10.0 x Appendix D x Appendix G x 1.0 Introduction Purpose and Scope 1.1 Sources of Safety Information 1.2 2.0 Regulatory Requirements 3.0 Roles and Responsibilities Research Safety Oversight Committee (RSOC) 3.1 Campus-Wide Chemical Safety Committee (CCSC) 3.1 Other Safety Committees 3.2 Office of Environmental Health & Safety 3.2 Principal Investigator (PI) 3.3 Training Requirements 3.5 4.0 Basics of Laboratory Safety Hazard, Risk, and Safety Management 4.1 Hierarchy of Safety Controls 4.1 Group Safety Management and Safety Culture 4.4 USC Chemical Hygiene Plan iii Revised 03/2021 Basics of Lab Facilities, Equipment, and Emergency Supplies 4.7 Emergency Equipment and Supplies 4.15 Open Flames 4.20 5.0 Hazard Communication Labeling and Signage Systems 5.2 Labelling and Signage in the Lab: What You Need to Do 5.5 Safety Data Sheets (SDSs): What Are They? 5.7 SDSs in The Lab: What You Need to Do 5.8 6.0 Hazardous Chemicals and Hazard Classification Introduction 6.1 Health-Hazardous Chemicals: Routes of Exposure 6.2 Particularly Hazardous Substances (PHS) 6.16 7.0 Chemical -

THE MONATOMIC IONS! 1. What Is the Formula for Silver? Ag 2. What Is
Name: ______________________________ THE MONATOMIC IONS! 1. What is the formula for silver? Ag+ 22. What is the formula for cobalt (II)? Co2+ 2. What is the formula for cadmium? Cd2+ 23. What is the formula for chromium (II)? Cr2+ 3. What is the formula for manganese (II)? Mn2+ 24. What is the formula for copper (II)? Cu2+ 4. What is the formula for nickel (II)? Ni2+ 25. What is the formula for tin (IV)? Sn4+ 5. What is the formula for chromous? Cr2+ 26. What is the formula for lead (IV)? Pb4+ 6. What is the formula for zinc? Zn2+ 27. What is the formula for iron (III)? Fe3+ 2+ 2+ 7. What is the formula for cobaltous? Co 28. What is the formula for mercury (I)? Hg2 8. What is the formula for cuprous? Cu+ 29. What is the formula for lead (II)? Pb2+ 9. What is the formula for ferrous? Fe2+ 30. What is the formula for mercury (II)? Hg2+ 2+ 2+ 10. What is the formula for mercurous? Hg2 31. What is the formula for iron (II)? Fe 11. What is the formula for stannous? Sn2+ 32. What is the formula for copper (I)? Cu+ 12. What is the formula for plumbous? Pb2+ 33. What is the formula for tin (II)? Sn2+ 13. What is the formula for chromic? Cr3+ 34. What is the formula for fluoride? F- 14. What is the formula for cobaltic? Co3+ 35. What is the formula for chloride? Cl- 15. What is the formula for cupric? Cu2+ 36. What is the formula for hydride? H- 16. -

Compound Name
Indicate Type of Compound: Compound Name Write your answer here I = ionic, A= acid, M = molecular manganese (II) bromite I Mn(BrO2)2 manganese (II) phosphite I Mn3(PO3)2 rubidium sulfite I Rb2SO3 hydroselenic acid A H2Se(aq) sodium perbromate I NaBrO4 cobalt (III) chromate I Co2(CrO4)3 antimony (V) nitrite I Sb(NO2)5 chloric acid A HClO3(aq) pentaselenium decabromide M Se5Br10 disulfur decachloride M S2Cl10 nickel (III) nitrate I Ni(NO3)3 copper (II) bromide I CuBr2 nickel (II) hydrogen phosphate I NiHPO4 iron (II) hydrogen sulfate I Fe(HSO4)2 bismuth (V) acetate I Bi(C2H3O2)5 sulfurous acid A H2SO3(aq) sulfuric acid A H2SO4(aq) nickel (II) chloride I NiCl2 tin (IV) phosphate I Sn3(PO4)4 mercury (I) iodate I Hg2(IO3)2 Compound Indicate Type of Compound: Write your answer here Formula I = ionic, A= acid, M = molecular Co(HCO3)2 I (with VOS metal) cobalt (II) hydrogen carbonate Cs2S I cesium sulfide Ca(IO2)2 I calcium iodite Ba2C I barium carbide Mn(CO3)2 I (with VOS metal) manganese (IV) carbonate CuBrO2 I (with VOS metal) copper (I) bromite AgHS I silver hydrogen sulfide C9N10 M nonacarbon decanitride CrI2 I (with VOS metal) chromium (II) iodide Mg(NO3)2 I magnesium nitrate HC2H3O2 (aq) A acetic acid HClO2 (aq) A chlorous acid Be(IO4)2 I beryllium periodate HIO4(aq) A periodic acid BaO I barium oxide Cd(BrO3)2 I cadmium bromate Bi(CN)5 I (with VOS metal) bismuth (V) cyanide AuHS I (with VOS metal) gold (I) hydrogen sulfide AuClO I (with VOS metal) gold (I) hypochlorite Na2CO3 I sodium carbonate Indicate Type of Compound: Compound Name -

2013-‐08-‐04 1 Oxidation and Reduction
2013-08-04 Rules for Assigning B) Oxidation State Determination Oxidation State (O.S.)* 1. O.S. of an atom in pure element = 0 Oxidation and Reduction For an atom, electrons can be lost or gained during a 2. O.S. of monoatomic ion = charge chemical reaction. 3. O.S. of Oxygen (O) is -2 in most of its covalent and ionic compounds (-1 in peroxides). If electrons are lost: OXIDATION The state becomes more positive 4. O.S. of H is +1 when combined with a non-metal 5. O.S. of H is -1 when combined with a metal If electrons are gained: REDUCTION The state becomes more negative 6. O.S. of Fluorine (F) is -1 in compounds 7. The sum of O.S. is 0 in compounds Oxidation State (O.S.) or Oxidation Number (O.N.) Number of electrons lost or gained during a chemical 8. The sum of O.S. equals charge in ions change * A compilation from different textbooks Elements with Fixed Oxidation States (O.S.)* Instead of memorizing elements that show different O.S., remember those that have a fixed one: Group 1 (IA) elements: Always +1 Group 2 (IIA) elements: Always +2 Aluminum, Al, and Scandium, Sc: Always +3 Zinc, Zn, and Cadmium, Cd: Always +2 Silver, Ag: Always +1 * A compilation from different textbooks 1 2013-08-04 OXIDATION STATE DETERMINATION EXERCISES Assign oxidation state (O.S.) to each element in the following compounds: a) SO3 i) CrCl3 2- 2- b) SO3 j) CrO4 2- c) KMnO4 k) Cr2O7 d) MnO2 l) HClO4 e) HNO3 m) HClO3 f) S8 n) HClO2 g) CH2Cl2 o) HClO h) SCl2 p) K2SO4 2 2013-08-04 C) A Nomenclature Proposal MORE GENERAL OBSERVATIONS Problematic of Nomenclature Coverage in Textbooks • Flow charts of various complexities (relatively often!) Flow charts, rules, … and tables, tables, tables … (Memorization!) ü From 3-box flow charts to 14-box ones GENERAL OBSERVATIONS • Link with acid/base concepts (sometimes, but rarely!) • Appears in chapters 2 or 3 of textbooks (often!) ü So, it is to be covered “at once”; as a single topic. -

Principles of Chemical Nomenclature a GUIDE to IUPAC RECOMMENDATIONS Principles of Chemical Nomenclature a GUIDE to IUPAC RECOMMENDATIONS
Principles of Chemical Nomenclature A GUIDE TO IUPAC RECOMMENDATIONS Principles of Chemical Nomenclature A GUIDE TO IUPAC RECOMMENDATIONS G.J. LEIGH OBE TheSchool of Chemistry, Physics and Environmental Science, University of Sussex, Brighton, UK H.A. FAVRE Université de Montréal Montréal, Canada W.V. METANOMSKI Chemical Abstracts Service Columbus, Ohio, USA Edited by G.J. Leigh b Blackwell Science © 1998 by DISTRIBUTORS BlackweilScience Ltd Marston Book Services Ltd Editorial Offices: P0 Box 269 Osney Mead, Oxford 0X2 0EL Abingdon 25 John Street, London WC1N 2BL Oxon 0X14 4YN 23 Ainslie Place, Edinburgh EH3 6AJ (Orders:Tel:01235 465500 350 Main Street, Maiden Fax: MA 02 148-5018, USA 01235 465555) 54 University Street, Carlton USA Victoria 3053, Australia BlackwellScience, Inc. 10, Rue Casmir Delavigne Commerce Place 75006 Paris, France 350 Main Street Malden, MA 02 148-5018 Other Editorial Offices: (Orders:Tel:800 759 6102 Blackwell Wissenschafts-Verlag GmbH 781 388 8250 KurfUrstendamm 57 Fax:781 388 8255) 10707 Berlin, Germany Canada Blackwell Science KK Copp Clark Professional MG Kodenmacho Building 200Adelaide St West, 3rd Floor 7—10 Kodenmacho Nihombashi Toronto, Ontario M5H 1W7 Chuo-ku, Tokyo 104, Japan (Orders:Tel:416 597-1616 800 815-9417 All rights reserved. No part of Fax:416 597-1617) this publication may be reproduced, stored in a retrieval system, or Australia BlackwellScience Pty Ltd transmitted, in any form or by any 54 University Street means, electronic, mechanical, Carlton, Victoria 3053 photocopying, recording or otherwise, (Orders:Tel:39347 0300 except as permitted by the UK Fax:3 9347 5001) Copyright, Designs and Patents Act 1988, without the prior permission of the copyright owner. -

HYSYS OLI Interface
HYSYS® 2004.2 OLI Interface Reference Guide Copyright October 2005 Copyright © 1981-2005 by Aspen Technology, Inc. All rights reserved. Aspen Accounting.21™, Aspen ACM Model Export, Aspen ACOL™, Aspen ACX™ Upgrade to ACOL™, Aspen Adsim®, Aspen Advisor™, Aspen Aerotran®, Aspen Alarm & Event™, Aspen APLE™, Aspen Apollo™, Aspen AtOMS™, Aspen Batch and Event Extractor, Aspen Batch Plus®, Aspen Batch.21™, Aspen Batch.21™ CBT, Aspen BatchCAD™, Aspen BatchSep™, Aspen Blend Model Library™, Aspen Blend™, Aspen BP Crude Oil Database, Aspen Calc CBT, Aspen Calc™, Aspen Capable-to-Promise®, Aspen CatRef®, Aspen Chromatography®, Aspen Cim-IO Core™, Aspen Cim-IO™ for @AGlance, Aspen Cim-IO™ for ABB 1180/ 1190 via DIU, Aspen Cim-IO™ for Bailey SemAPI, Aspen Cim-IO™ for DDE, Aspen Cim-IO™ for Eurotherm Gauge via DCP, Aspen Cim-IO™ for Fisher-Rosemount Chip, Aspen Cim-IO™ for Fisher-Rosemount RNI, Aspen Cim-IO™ for Foxboro FOXAPI, Aspen Cim-IO™ for G2, Aspen Cim-IO™ for GE FANUC via HCT, Aspen Cim-IO™ for Hitachi Ex Series, Aspen Cim-IO™ for Honeywell TDC 3000 via HTL/access, Aspen Cim-IO™ for Intellution Fix, Aspen Cim-IO™ for Measurex MCN, Aspen Cim-IO™ for Measurex ODX, Aspen Cim-IO™ for Moore Apacs via Nim (RNI), Aspen Cim-IO™ for OPC, Aspen Cim-IO™ for PI, Aspen Cim- IO™ for RSLinx, Aspen Cim-IO™ for SetCim/InfoPlus-X/InfoPlus.21, Aspen Cim-IO™ for Toshiba Tosdic, Aspen Cim-IO™ for ULMA 3D, Aspen Cim-IO™ for Westinghouse, Aspen Cim-IO™ for WonderWare InTouch, Aspen Cim-IO™ for Yokogawa ACG10S, Aspen Cim-IO™ for Yokogawa EW3, Aspen Collaborative Forecasting™, -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G. -

Introduction to Nomenclature
INTRODUCTION TO NOMENCLATURE 1. Binary ionic compounds - one metal element & one non-metal. i.e. NaCl & TiH2 Separate the compound into ions before naming it. i.e Na+ Cl- Ti2+ H- H- The sum of the positive and negative charges must = 0. Some elements like Ti may have different positive charges which may be calculated from the negative charges which are fixed. (Refer to table, next page). The cation is named first, then the anion, i.e. sodium chloride and titanium (II) hydride. The Roman numeral on the name of the cation is necessary to specify the charge if the element is one that can have different charges. Na can only have a +1 charge so it needs no Roman numerals. Ti can have charges of 2+ or 3+ or 4+ so Roman numerals are needed. 2. Binary molecular compounds are composed of two non-metals and no ions are present. i.e. CO CO2 N2O4 PCl3 CF4 IF7 Use Greek numerical prefixes to designate the number of atoms in the molecule: i.e carbon monoxide, carbon dioxide, dinitrogen tetroxide, etc. Note – if the first atom written is singular then the mono prefix is not used – but if the second atom is singular – then the mono prefix MUST be used in the name. The second atom written has the - ide suffix, even though it is not a “true” anion. 1-mono, 2-di, 3-tri , 4-tetra, 5-penta, 6-hexa , 7-hepta , 8-octa , 9-nona, 10-deca 3. Compounds with oxyanions are named, with the cation first, then the anion. -
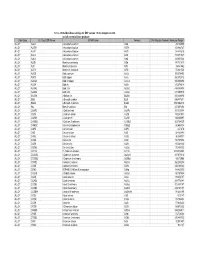
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212