Purification, Properties, and Kinetics of D-Ribulokinase from Aerobacter Aerogenes " (1980)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Atpase Domain Common to Prokaryotic Cell Cycle Proteins
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 7290-7294, August 1992 Biochemistry An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp7O heat shock proteins (structural comparison/property pattern/remote homology) PEER BORK, CHRIS SANDER, AND ALFONSO VALENCIA European Molecular Biology Laboratory, D-6900 Heidelberg, Federal Republic of Germany Communicated by Russell F. Doolittle, March 6, 1992 ABSTRACT The functionally diverse actin, hexokinase, and hsp7O protein families have in common an ATPase domain of known three-dimensional structure. Optimal superposition ofthe three structures and alignment ofmany sequences in each of the three families has revealed a set of common conserved residues, distributed in five sequence motifs, which are in- volved in ATP binding and in a putative interdomain hinge. From the multiple sequence aliment in these motifs a pattern of amino acid properties required at each position is defined. The discriminatory power of the pattern is in part due to the use of several known three-dimensional structures and many sequences and in part to the "property" method ofgeneralizing from observed amino acid frequencies to amino acid fitness at each sequence position. A sequence data base search with the pattern significantly matches sugar kinases, such as fuco-, glucono-, xylulo-, ribulo-, and glycerokinase, as well as the prokaryotic cell cycle proteins MreB, FtsA, and StbA. These are predicted to have subdomains with the same tertiary structure as the ATPase subdomains Ia and Ha of hexokinase, actin, and Hsc7O, a very similar ATP binding pocket, and the capacity for interdomain hinge motion accompanying func- tional state changes. -

Non-Homologous Isofunctional Enzymes: a Systematic Analysis Of
Omelchenko et al. Biology Direct 2010, 5:31 http://www.biology-direct.com/content/5/1/31 RESEARCH Open Access Non-homologousResearch isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution Marina V Omelchenko, Michael Y Galperin*, Yuri I Wolf and Eugene V Koonin Abstract Background: Evolutionarily unrelated proteins that catalyze the same biochemical reactions are often referred to as analogous - as opposed to homologous - enzymes. The existence of numerous alternative, non-homologous enzyme isoforms presents an interesting evolutionary problem; it also complicates genome-based reconstruction of the metabolic pathways in a variety of organisms. In 1998, a systematic search for analogous enzymes resulted in the identification of 105 Enzyme Commission (EC) numbers that included two or more proteins without detectable sequence similarity to each other, including 34 EC nodes where proteins were known (or predicted) to have distinct structural folds, indicating independent evolutionary origins. In the past 12 years, many putative non-homologous isofunctional enzymes were identified in newly sequenced genomes. In addition, efforts in structural genomics resulted in a vastly improved structural coverage of proteomes, providing for definitive assessment of (non)homologous relationships between proteins. Results: We report the results of a comprehensive search for non-homologous isofunctional enzymes (NISE) that yielded 185 EC nodes with two or more experimentally characterized - or predicted - structurally unrelated proteins. Of these NISE sets, only 74 were from the original 1998 list. Structural assignments of the NISE show over-representation of proteins with the TIM barrel fold and the nucleotide-binding Rossmann fold. From the functional perspective, the set of NISE is enriched in hydrolases, particularly carbohydrate hydrolases, and in enzymes involved in defense against oxidative stress. -

Recent Advances in Trypanosomatid Research: Genome Organization, Expression, Metabolism, Taxonomy and Evolution
Parasitology Recent advances in trypanosomatid research: genome organization, expression, metabolism, cambridge.org/par taxonomy and evolution 1 2 3,4 5,6 Review Dmitri A. Maslov , Fred R. Opperdoes , Alexei Y. Kostygov , Hassan Hashimi , Julius Lukeš5,6 and Vyacheslav Yurchenko3,5,7 Cite this article: Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V 1Department of Molecular, Cell and Systems Biology, University of California – Riverside, Riverside, California, USA; (2018). Recent advances in trypanosomatid 2de Duve Institute, Université Catholique de Louvain, Brussels, Belgium; 3Life Science Research Centre, Faculty of research: genome organization, expression, 4 metabolism, taxonomy and evolution. Science, University of Ostrava, Ostrava, Czech Republic; Zoological Institute of the Russian Academy of Sciences, 5 Parasitology 1–27. https://doi.org/10.1017/ St. Petersburg, Russia; Biology Centre, Institute of Parasitology, Czech Academy of Sciences, České Budejovice 6 S0031182018000951 (Budweis), Czech Republic; University of South Bohemia, Faculty of Sciences, České Budejovice (Budweis), Czech Republic and 7Martsinovsky Institute of Medical Parasitology, Tropical and Vector Borne Diseases, Sechenov Received: 30 January 2018 University, Moscow, Russia Revised: 23 April 2018 Accepted: 23 April 2018 Abstract Key words: Unicellular flagellates of the family Trypanosomatidae are obligatory parasites of inverte- Gene exchange; kinetoplast; metabolism; molecular and cell biology; taxonomy; brates, vertebrates and plants. Dixenous species are aetiological agents of a number of diseases trypanosomatidae in humans, domestic animals and plants. Their monoxenous relatives are restricted to insects. Because of the high biological diversity, adaptability to dramatically different environmental Author for correspondence: conditions, and omnipresence, these protists have major impact on all biotic communities Vyacheslav Yurchenko, E-mail: vyacheslav. -
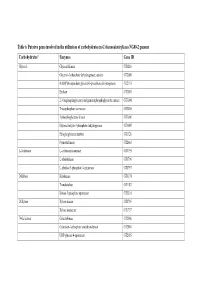
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

A Global Analysis of Enzyme Compartmentalization to Glycosomes
pathogens Article A Global Analysis of Enzyme Compartmentalization to Glycosomes Hina Durrani 1, Marshall Hampton 2 , Jon N. Rumbley 3 and Sara L. Zimmer 1,* 1 Department of Biomedical Sciences, University of Minnesota Medical School, Duluth Campus, Duluth, MN 55812, USA; [email protected] 2 Mathematics & Statistics Department, University of Minnesota Duluth, Duluth, MN 55812, USA; [email protected] 3 College of Pharmacy, University of Minnesota, Duluth Campus, Duluth, MN 55812, USA; [email protected] * Correspondence: [email protected] Received: 25 March 2020; Accepted: 9 April 2020; Published: 12 April 2020 Abstract: In kinetoplastids, the first seven steps of glycolysis are compartmentalized into a glycosome along with parts of other metabolic pathways. This organelle shares a common ancestor with the better-understood eukaryotic peroxisome. Much of our understanding of the emergence, evolution, and maintenance of glycosomes is limited to explorations of the dixenous parasites, including the enzymatic contents of the organelle. Our objective was to determine the extent that we could leverage existing studies in model kinetoplastids to determine the composition of glycosomes in species lacking evidence of experimental localization. These include diverse monoxenous species and dixenous species with very different hosts. For many of these, genome or transcriptome sequences are available. Our approach initiated with a meta-analysis of existing studies to generate a subset of enzymes with highest evidence of glycosome localization. From this dataset we extracted the best possible glycosome signal peptide identification scheme for in silico identification of glycosomal proteins from any kinetoplastid species. Validation suggested that a high glycosome localization score from our algorithm would be indicative of a glycosomal protein. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Transaldolasewglucose6phosphate Isomerase Bifunctional Enzyme and Ribulokinase As Factors to Increase Xylitol Production
Biosci. Biotechnol. Biochem., 67 (12), 25242532, 2003 TransaldolaseWGlucose6phosphate Isomerase Bifunctional Enzyme and Ribulokinase as Factors to Increase Xylitol Production from DArabitol in Gluconobacter oxydans Masakazu SUGIYAMA,õ Shunichi SUZUKI,NaotoTONOUCHI,andKenzoYOKOZEKI AminoScience Laboratories, Ajinomoto Co., Inc., Suzukicho, Kawasakiku, Kawasakishi 2108681, Japan Received May 20, 2003; Accepted September 8, 2003 Xylitol production from Darabitol by the membrane and soluble fractions of Gluconobacter oxydans was investigated. Two proteins in the soluble fraction were found to have the ability to increase xylitol production. Both of these xylitolincreasing factors were puried, and on the basis of their NH2terminal amino acid sequences the genes encoding both of the factors were cloned. Expression of the cloned genes in Escherichia Fig. 1. Schematic Representation of Xylitol Production from coli showed that one of the xylitolincreasing factors DArabitol by G. oxydans. is the bifunctional enzyme transaldolaseWglucose AraDH, membranebound Darabitol dehydrogenase; XDH, 6phosphate isomerase, and the other is ribulokinase. soluble NADHdependent xylitol dehydrogenase. Using membrane and soluble fractions of G. oxydans, 3.8 gWl of xylitol were produced from 10 gWl Darabitol after incubation for 40 h, and addition of puried for producing xylitol from Darabitol, thereby lower recombinant transaldolaseWglucose6phosphate isom ing manufacturing costs by allowing xylitol to be erase or ribulokinase increased xylitol -

Supporting Information
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2019 Supporting information for Metabolomic and proteomic changes induced by growth inhibitory concentrations of copper in the biofilm-forming marine bacterium Pseudoalteromonas lipolytica Laurie Favre,a Annick Ortalo-Magné,a,* Lionel Kerloch,a Carole Pichereaux,b,c Benjamin Misson,d Jean-François Brianda, Cédric Garnierd and Gérald Culiolia,* a Université de Toulon, MAPIEM EA 4323, Toulon, France. b Fédération de Recherche FR3450, Agrobiosciences, Interaction et Biodiversité (AIB), CNRS, Toulouse, France.. c Institut de Pharmacologie et de Biologie Structurale, IPBS, Université de Toulouse, CNRS, UPS, Toulouse, France. d Univ de Toulon, Aix Marseille Univ, CNRS, IRD, MIO UM 110, Mediterranean Institute of Oceanography, La Garde, France. Detailed description of the experimental protocol used for Cu chemical speciation. Differential pulse anodic striping voltammetry (DPASV) measurements were performed to assess Cu chemical speciation in both VNSS and MB medium. All voltammetric measurements were performed using a PGSTAT12 potentiostat (EcoChemie, The Netherlands) equipped with a Metrohm 663 VA stand (Metrohm, Switzerland) on an Hg drop electrode. Analysis of the voltammetric peaks' (after 2nd derivative transformation) and construction of the pseudo- polarograms were performed automatically using ECDSoft software. The experimental procedure adapted from Nicolau et al. (2008) and Bravin et al. (2012) is explained briefly below. Pseudo-polarographic measurements (DPASV measurement with Edep from -1.0 to 0.0V by steps of 100 mV, using a deposition time of 30s) were initially performed on UV-irradiated seawater (collected outside the Toulon bay: ambient Cu concentration of 2.5 nM) at pH < 2 (by adding HNO3 Suprapur grade, Merck), to determine the deposition potential (Edep -0.35V) corresponding to the electrochemically "labile" Cu fraction (sum of free Cu ions plus inorganically bound Cu) (Figure S2). -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA