An Atpase Domain Common to Prokaryotic Cell Cycle Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fluorinated Mannosides Inhibit Cellular Fucosylation
Fluorinated mannosides inhibit cellular fucosylation. Johan F.A. Pijnenborg[a],†, Emiel Rossing[a],†, Marek Noga[b], Willem Titulaer[a], Raisa Veizaj[c], Dirk J. Lefeber[b,c] and Thomas J. Boltje*[a] [a] J.F.A. Pijnenborg, E. Rossing, W. Titulaer, Dr. T.J. Boltje Department of Synthetic Organic Chemistry Institute for Molecules and Materials, Radboud University Heyendaalseweg 135, 6525AJ, Nijmegen, The Netherlands [email protected] [b] Dr. M. Noga, Prof. D.J. Lefeber Department of Laboratory Medicine, Translational Metabolic Laboratory Radboud Institute for Molecular Life Sciences, Radboud University Medical Center Geert Grooteplein Zuid 10, 6525GA, Nijmegen, The Netherlands [c] R. Veizaj, Prof. D.J. Lefeber Department of Neurology Donders Institute for Brain, Cognition and Behavior, Radboud University Medical Center Geert Grooteplein Zuid 10, 6525GA, Nijmegen, The Netherlands [†] These authors contributed equally to this work. Supporting information for this article is given via a link at the end of the document. Abstract: Fucose sugars are expressed on mammalian cell L-Fucose (Fuc) is a 6-deoxyhexose expressed at the termini of glycan membranes as part of glycoconjugates and mediates essential chains that decorate cell surface proteins and lipids.1 The fucose physiological processes. The aberrant expression of fucosylated residues on glycoconjugates are essential mediators of physiological glycans has been linked to pathologies such as cancer, inflammation, processes. For example, the fucose moiety in the tetrasaccharide infection, and genetic disorders. Tools to modulate fucose expression sialyl Lewisx (sLex) expressed on leukocytes is recognized by selectin on living cells are needed to elucidate the biological role of fucose receptors that regulate leukocyte recruitment and extravasation. -

Non-Homologous Isofunctional Enzymes: a Systematic Analysis Of
Omelchenko et al. Biology Direct 2010, 5:31 http://www.biology-direct.com/content/5/1/31 RESEARCH Open Access Non-homologousResearch isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution Marina V Omelchenko, Michael Y Galperin*, Yuri I Wolf and Eugene V Koonin Abstract Background: Evolutionarily unrelated proteins that catalyze the same biochemical reactions are often referred to as analogous - as opposed to homologous - enzymes. The existence of numerous alternative, non-homologous enzyme isoforms presents an interesting evolutionary problem; it also complicates genome-based reconstruction of the metabolic pathways in a variety of organisms. In 1998, a systematic search for analogous enzymes resulted in the identification of 105 Enzyme Commission (EC) numbers that included two or more proteins without detectable sequence similarity to each other, including 34 EC nodes where proteins were known (or predicted) to have distinct structural folds, indicating independent evolutionary origins. In the past 12 years, many putative non-homologous isofunctional enzymes were identified in newly sequenced genomes. In addition, efforts in structural genomics resulted in a vastly improved structural coverage of proteomes, providing for definitive assessment of (non)homologous relationships between proteins. Results: We report the results of a comprehensive search for non-homologous isofunctional enzymes (NISE) that yielded 185 EC nodes with two or more experimentally characterized - or predicted - structurally unrelated proteins. Of these NISE sets, only 74 were from the original 1998 list. Structural assignments of the NISE show over-representation of proteins with the TIM barrel fold and the nucleotide-binding Rossmann fold. From the functional perspective, the set of NISE is enriched in hydrolases, particularly carbohydrate hydrolases, and in enzymes involved in defense against oxidative stress. -

Supplementary Material Gram-Scale Production of Sugar Nucleotides And
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is © The Royal Society of Chemistry 2021 Supplementary Material Gram-scale production of sugar nucleotides and their derivatives Shuang Li[a]#, Shuaishuai Wang[b]#, Yaqian Wang[a], Jingyao Qu[c], Xian-wei Liu[a], Peng George Wang[d], and Junqiang Fang*[a] [a] Junqiang Fang, Shuang Li, Yaqian Wang, Xian-wei Liu National Glycoengineering Research Center, Shandong Provincial Key Laboratory of Glycochemistry and Glycobiology, Shandong University, Qingdao, Shandong 266237, People’s Republic of China *Email: [email protected] [b] Shuaishuai Wang Department of Chemistry, George State University, Atlanta, GA, 30302-4098, US [c] Jingyao Qu State Key Laboratory of Microbial Technology, Shandong University, Qingdao, Shandong 266237, People’s Republic of China [d] Peng George Wang School of Medicine, Southern University of Science and Technology, Shenzhen, Guangdong 518055, People’s Republic of China # These authors contributed equally to this paper. I. Supplementary Figures..................................................................................................................................1 Figure S1. Effects of GlcNAc substrate concentration on conversion rate of UDP-GlcNAc......................1 Figure S2. Effect of buffer sytem on enzymatic conversion rate of UDP-GlcNAc.....................................1 Figure S3. Evaluation of recovery and recyclability of enzymes for UDP-GlcNAc...................................1 Table S1. Enzymes used in this work -

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

Table S1. List of Oligonucleotide Primers Used
Table S1. List of oligonucleotide primers used. Cla4 LF-5' GTAGGATCCGCTCTGTCAAGCCTCCGACC M629Arev CCTCCCTCCATGTACTCcgcGATGACCCAgAGCTCGTTG M629Afwd CAACGAGCTcTGGGTCATCgcgGAGTACATGGAGGGAGG LF-3' GTAGGCCATCTAGGCCGCAATCTCGTCAAGTAAAGTCG RF-5' GTAGGCCTGAGTGGCCCGAGATTGCAACGTGTAACC RF-3' GTAGGATCCCGTACGCTGCGATCGCTTGC Ukc1 LF-5' GCAATATTATGTCTACTTTGAGCG M398Arev CCGCCGGGCAAgAAtTCcgcGAGAAGGTACAGATACGc M398Afwd gCGTATCTGTACCTTCTCgcgGAaTTcTTGCCCGGCGG LF-3' GAGGCCATCTAGGCCATTTACGATGGCAGACAAAGG RF-5' GTGGCCTGAGTGGCCATTGGTTTGGGCGAATGGC RF-3' GCAATATTCGTACGTCAACAGCGCG Nrc2 LF-5' GCAATATTTCGAAAAGGGTCGTTCC M454Grev GCCACCCATGCAGTAcTCgccGCAGAGGTAGAGGTAATC M454Gfwd GATTACCTCTACCTCTGCggcGAgTACTGCATGGGTGGC LF-3' GAGGCCATCTAGGCCGACGAGTGAAGCTTTCGAGCG RF-5' GAGGCCTGAGTGGCCTAAGCATCTTGGCTTCTGC RF-3' GCAATATTCGGTCAACGCTTTTCAGATACC Ipl1 LF-5' GTCAATATTCTACTTTGTGAAGACGCTGC M629Arev GCTCCCCACGACCAGCgAATTCGATagcGAGGAAGACTCGGCCCTCATC M629Afwd GATGAGGGCCGAGTCTTCCTCgctATCGAATTcGCTGGTCGTGGGGAGC LF-3' TGAGGCCATCTAGGCCGGTGCCTTAGATTCCGTATAGC RF-5' CATGGCCTGAGTGGCCGATTCTTCTTCTGTCATCGAC RF-3' GACAATATTGCTGACCTTGTCTACTTGG Ire1 LF-5' GCAATATTAAAGCACAACTCAACGC D1014Arev CCGTAGCCAAGCACCTCGgCCGAtATcGTGAGCGAAG D1014Afwd CTTCGCTCACgATaTCGGcCGAGGTGCTTGGCTACGG LF-3' GAGGCCATCTAGGCCAACTGGGCAAAGGAGATGGA RF-5' GAGGCCTGAGTGGCCGTGCGCCTGTGTATCTCTTTG RF-3' GCAATATTGGCCATCTGAGGGCTGAC Kin28 LF-5' GACAATATTCATCTTTCACCCTTCCAAAG L94Arev TGATGAGTGCTTCTAGATTGGTGTCggcGAAcTCgAGCACCAGGTTG L94Afwd CAACCTGGTGCTcGAgTTCgccGACACCAATCTAGAAGCACTCATCA LF-3' TGAGGCCATCTAGGCCCACAGAGATCCGCTTTAATGC RF-5' CATGGCCTGAGTGGCCAGGGCTAGTACGACCTCG -

The Role of the Salvage Pathway in Nucleotide Sugar Biosynthesis
THE ROLE OF THE SALVAGE PATHWAY IN NUCLEOTIDE SUGAR BIOSYNTHESIS: IDENTIFICATION OF SUGAR KINASES AND NDP-SUGAR PYROPHOSPHORYLASES by TING YANG (Under the Direction of Maor Bar-Peled) ABSTRACT The synthesis of polysaccharides, glycoproteins, glycolipids, glycosylated secondary metabolites and hormones requires a large number of glycosyltransferases and a constant supply of nucleotide sugars. In plants, photosynthesis and the NDP-sugar inter-conversion pathway are the major entry points to form NDP-sugars. In addition to these pathways is the salvage pathway, a less understood metabolism that provides the flux of NDP-sugars. This latter pathway involves the hydrolysis of glycans to free sugars, sugar transport, sugar phosphorylation and nucleotidylation. The balance between glycan synthesis and recycling as well as its regulation at various plant developmental stages remains elusive as many of the molecular components are unknown. To understand how the salvage pathway contributes to the sugar flux and cell wall biosynthesis, my research focused on the functional identification of salvage pathway sugar kinases and NDP-sugar pyrophosphorylases. This research led to the first identification and enzymatic characterization of galacturonic acid kinase (GalA kinase), galactokinase (GalK), a broad UDP-sugar pyrophosphorylase (sloppy), two promiscuous UDP-GlcNAc pyrophosphorylases (GlcNAc-1-P uridylyltransferases), as well as UDP-sugar pyrophosphorylase paralogs from Trypanosoma cruzi and Leishmania major. To evaluate the salvage pathway in plant biology, we further investigated a sugar kinase mutant: galacturonic acid kinase mutant (galak) and determined if and how galak KO mutant affects the synthesis of glycans in Arabidopsis. Feeding galacturonic acid to the seedlings exhibited a 40-fold accumulation of free GalA in galak mutant, while the wild type (WT) plant readily metabolizes the fed-sugar. -

Recent Advances in Trypanosomatid Research: Genome Organization, Expression, Metabolism, Taxonomy and Evolution
Parasitology Recent advances in trypanosomatid research: genome organization, expression, metabolism, cambridge.org/par taxonomy and evolution 1 2 3,4 5,6 Review Dmitri A. Maslov , Fred R. Opperdoes , Alexei Y. Kostygov , Hassan Hashimi , Julius Lukeš5,6 and Vyacheslav Yurchenko3,5,7 Cite this article: Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V 1Department of Molecular, Cell and Systems Biology, University of California – Riverside, Riverside, California, USA; (2018). Recent advances in trypanosomatid 2de Duve Institute, Université Catholique de Louvain, Brussels, Belgium; 3Life Science Research Centre, Faculty of research: genome organization, expression, 4 metabolism, taxonomy and evolution. Science, University of Ostrava, Ostrava, Czech Republic; Zoological Institute of the Russian Academy of Sciences, 5 Parasitology 1–27. https://doi.org/10.1017/ St. Petersburg, Russia; Biology Centre, Institute of Parasitology, Czech Academy of Sciences, České Budejovice 6 S0031182018000951 (Budweis), Czech Republic; University of South Bohemia, Faculty of Sciences, České Budejovice (Budweis), Czech Republic and 7Martsinovsky Institute of Medical Parasitology, Tropical and Vector Borne Diseases, Sechenov Received: 30 January 2018 University, Moscow, Russia Revised: 23 April 2018 Accepted: 23 April 2018 Abstract Key words: Unicellular flagellates of the family Trypanosomatidae are obligatory parasites of inverte- Gene exchange; kinetoplast; metabolism; molecular and cell biology; taxonomy; brates, vertebrates and plants. Dixenous species are aetiological agents of a number of diseases trypanosomatidae in humans, domestic animals and plants. Their monoxenous relatives are restricted to insects. Because of the high biological diversity, adaptability to dramatically different environmental Author for correspondence: conditions, and omnipresence, these protists have major impact on all biotic communities Vyacheslav Yurchenko, E-mail: vyacheslav. -
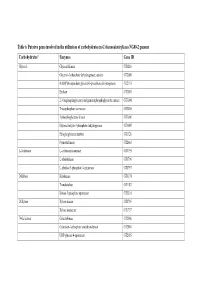
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

A Global Analysis of Enzyme Compartmentalization to Glycosomes
pathogens Article A Global Analysis of Enzyme Compartmentalization to Glycosomes Hina Durrani 1, Marshall Hampton 2 , Jon N. Rumbley 3 and Sara L. Zimmer 1,* 1 Department of Biomedical Sciences, University of Minnesota Medical School, Duluth Campus, Duluth, MN 55812, USA; [email protected] 2 Mathematics & Statistics Department, University of Minnesota Duluth, Duluth, MN 55812, USA; [email protected] 3 College of Pharmacy, University of Minnesota, Duluth Campus, Duluth, MN 55812, USA; [email protected] * Correspondence: [email protected] Received: 25 March 2020; Accepted: 9 April 2020; Published: 12 April 2020 Abstract: In kinetoplastids, the first seven steps of glycolysis are compartmentalized into a glycosome along with parts of other metabolic pathways. This organelle shares a common ancestor with the better-understood eukaryotic peroxisome. Much of our understanding of the emergence, evolution, and maintenance of glycosomes is limited to explorations of the dixenous parasites, including the enzymatic contents of the organelle. Our objective was to determine the extent that we could leverage existing studies in model kinetoplastids to determine the composition of glycosomes in species lacking evidence of experimental localization. These include diverse monoxenous species and dixenous species with very different hosts. For many of these, genome or transcriptome sequences are available. Our approach initiated with a meta-analysis of existing studies to generate a subset of enzymes with highest evidence of glycosome localization. From this dataset we extracted the best possible glycosome signal peptide identification scheme for in silico identification of glycosomal proteins from any kinetoplastid species. Validation suggested that a high glycosome localization score from our algorithm would be indicative of a glycosomal protein. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Engineering Yeast Hexokinase 2 for Improved Tolerance Toward Xylose-Induced Inactivation
Engineering Yeast Hexokinase 2 for Improved Tolerance Toward Xylose-Induced Inactivation Basti Bergdahl*, Anders G. Sandstro¨ m, Celina Borgstro¨ m, Tarinee Boonyawan¤, Ed W. J. van Niel, Marie F. Gorwa-Grauslund Division of Applied Microbiology, Department of Chemistry, Lund University, Lund, Sweden Abstract Hexokinase 2 (Hxk2p) from Saccharomyces cerevisiae is a bi-functional enzyme being both a catalyst and an important regulator in the glucose repression signal. In the presence of xylose Hxk2p is irreversibly inactivated through an autophosphorylation mechanism, affecting all functions. Consequently, the regulation of genes involved in sugar transport and fermentative metabolism is impaired. The aim of the study was to obtain new Hxk2p-variants, immune to the autophosphorylation, which potentially can restore the repressive capability closer to its nominal level. In this study we constructed the first condensed, rationally designed combinatorial library targeting the active-site in Hxk2p. We combined protein engineering and genetic engineering for efficient screening and identified a variant with Phe159 changed to tyrosine. This variant had 64% higher catalytic activity in the presence of xylose compared to the wild-type and is expected to be a key component for increasing the productivity of recombinant xylose-fermenting strains for bioethanol production from lignocellulosic feedstocks. Citation: Bergdahl B, Sandstro¨m AG, Borgstro¨m C, Boonyawan T, van Niel EWJ, et al. (2013) Engineering Yeast Hexokinase 2 for Improved Tolerance Toward Xylose-Induced Inactivation. PLoS ONE 8(9): e75055. doi:10.1371/journal.pone.0075055 Editor: Paul Hoskisson, University of Strathclyde, United States of America Received June 29, 2013; Accepted August 5, 2013; Published September 6, 2013 Copyright: ß 2013 Bergdahl et al.