Engineering Yeast Hexokinase 2 for Improved Tolerance Toward Xylose-Induced Inactivation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Atpase Domain Common to Prokaryotic Cell Cycle Proteins
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 7290-7294, August 1992 Biochemistry An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp7O heat shock proteins (structural comparison/property pattern/remote homology) PEER BORK, CHRIS SANDER, AND ALFONSO VALENCIA European Molecular Biology Laboratory, D-6900 Heidelberg, Federal Republic of Germany Communicated by Russell F. Doolittle, March 6, 1992 ABSTRACT The functionally diverse actin, hexokinase, and hsp7O protein families have in common an ATPase domain of known three-dimensional structure. Optimal superposition ofthe three structures and alignment ofmany sequences in each of the three families has revealed a set of common conserved residues, distributed in five sequence motifs, which are in- volved in ATP binding and in a putative interdomain hinge. From the multiple sequence aliment in these motifs a pattern of amino acid properties required at each position is defined. The discriminatory power of the pattern is in part due to the use of several known three-dimensional structures and many sequences and in part to the "property" method ofgeneralizing from observed amino acid frequencies to amino acid fitness at each sequence position. A sequence data base search with the pattern significantly matches sugar kinases, such as fuco-, glucono-, xylulo-, ribulo-, and glycerokinase, as well as the prokaryotic cell cycle proteins MreB, FtsA, and StbA. These are predicted to have subdomains with the same tertiary structure as the ATPase subdomains Ia and Ha of hexokinase, actin, and Hsc7O, a very similar ATP binding pocket, and the capacity for interdomain hinge motion accompanying func- tional state changes. -

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

Table S1. List of Oligonucleotide Primers Used
Table S1. List of oligonucleotide primers used. Cla4 LF-5' GTAGGATCCGCTCTGTCAAGCCTCCGACC M629Arev CCTCCCTCCATGTACTCcgcGATGACCCAgAGCTCGTTG M629Afwd CAACGAGCTcTGGGTCATCgcgGAGTACATGGAGGGAGG LF-3' GTAGGCCATCTAGGCCGCAATCTCGTCAAGTAAAGTCG RF-5' GTAGGCCTGAGTGGCCCGAGATTGCAACGTGTAACC RF-3' GTAGGATCCCGTACGCTGCGATCGCTTGC Ukc1 LF-5' GCAATATTATGTCTACTTTGAGCG M398Arev CCGCCGGGCAAgAAtTCcgcGAGAAGGTACAGATACGc M398Afwd gCGTATCTGTACCTTCTCgcgGAaTTcTTGCCCGGCGG LF-3' GAGGCCATCTAGGCCATTTACGATGGCAGACAAAGG RF-5' GTGGCCTGAGTGGCCATTGGTTTGGGCGAATGGC RF-3' GCAATATTCGTACGTCAACAGCGCG Nrc2 LF-5' GCAATATTTCGAAAAGGGTCGTTCC M454Grev GCCACCCATGCAGTAcTCgccGCAGAGGTAGAGGTAATC M454Gfwd GATTACCTCTACCTCTGCggcGAgTACTGCATGGGTGGC LF-3' GAGGCCATCTAGGCCGACGAGTGAAGCTTTCGAGCG RF-5' GAGGCCTGAGTGGCCTAAGCATCTTGGCTTCTGC RF-3' GCAATATTCGGTCAACGCTTTTCAGATACC Ipl1 LF-5' GTCAATATTCTACTTTGTGAAGACGCTGC M629Arev GCTCCCCACGACCAGCgAATTCGATagcGAGGAAGACTCGGCCCTCATC M629Afwd GATGAGGGCCGAGTCTTCCTCgctATCGAATTcGCTGGTCGTGGGGAGC LF-3' TGAGGCCATCTAGGCCGGTGCCTTAGATTCCGTATAGC RF-5' CATGGCCTGAGTGGCCGATTCTTCTTCTGTCATCGAC RF-3' GACAATATTGCTGACCTTGTCTACTTGG Ire1 LF-5' GCAATATTAAAGCACAACTCAACGC D1014Arev CCGTAGCCAAGCACCTCGgCCGAtATcGTGAGCGAAG D1014Afwd CTTCGCTCACgATaTCGGcCGAGGTGCTTGGCTACGG LF-3' GAGGCCATCTAGGCCAACTGGGCAAAGGAGATGGA RF-5' GAGGCCTGAGTGGCCGTGCGCCTGTGTATCTCTTTG RF-3' GCAATATTGGCCATCTGAGGGCTGAC Kin28 LF-5' GACAATATTCATCTTTCACCCTTCCAAAG L94Arev TGATGAGTGCTTCTAGATTGGTGTCggcGAAcTCgAGCACCAGGTTG L94Afwd CAACCTGGTGCTcGAgTTCgccGACACCAATCTAGAAGCACTCATCA LF-3' TGAGGCCATCTAGGCCCACAGAGATCCGCTTTAATGC RF-5' CATGGCCTGAGTGGCCAGGGCTAGTACGACCTCG -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Genetic Tools for High Yield Protein Production with Bacillus Megaterium
Genetic tools for high yield protein production with Bacillus megaterium Von der Fakultät für Lebenswissenschaften der Technischen Universität Carolo-Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigte D i s s e r t a t i o n von Simon Stammen aus Kevelaer 1. Referent: Professor Dr. Dieter Jahn 2. Referent: Professor Dr. Petra Dersch eingereicht am: 01.09.2010 mündliche Prüfung (Disputation) am: 08.10.2010 Druckjahr 2010 Vorveröffentlichungen der Dissertation: Teilergebnisse aus dieser Arbeit wurden mit Genehmigung der Fakultät für Lebenswissenschaften, vertreten durch den Mentor der Arbeit, in folgenden Beiträgen vorab veröffentlicht: Publikationen: Stammen, S. , Schuller, F., Dietrich, S., Gamer, M., Biedendieck R. and D. Jahn. 2010. Application of Escherichia coli phage K1E DNA-dependent RNA polymerase for in vitro RNA synthesis and in vivo protein production in Bacillus megaterium . Appl Microbiol Biotechnol 88 :529-539. Stammen, S. , Müller, B. K., Korneli, C., Biedendieck, R., Gamer, M., Franco-Lara, E. and D. Jahn. 2010. High yield intra- and extracellular protein production using Bacillus megaterium . Appl Environ Microbiol 76 :4037-4046. Bunk, B., Schulz, A., Stammen, S. , Münch, R., Warren, M. J., Rohde, M., Jahn, D. and R. Biedendieck. 2010. A short story about a big magic bug. Bioengineered Bugs 1:1-7. Gamer, M., Fröde, D., Biedendieck, R., Stammen, S. and D. Jahn. 2009. A T7 RNA polymerase-dependent gene expression system for Bacillus megaterium . Appl Microbiol Biotechnol 82 :1195-1203. Tagungs- und Industriebeiträge: Stammen, S. , Schuller, F., Dietrich, S., Gamer, M., Biedendieck, R. and D. Jahn. 2010. A novel DNA-dependent RNA polymerase of Escherichia coli phage K1E: production, characterization and application. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -

Regulation of Phosphotransferases in Glucose- and Xylose-Fermenting Yeasts
Copyright © 1997 by Humana Press Inc. All rights of any nature whatsoever reserved. 0273-2289/97/63-65–0097$11.00 Regulation of Phosphotransferases in Glucose- and Xylose-Fermenting Yeasts V INA W. YANG AND THOMAS W. JEFFRIES * Institute for Microbial and Biochemical Technology, Forest Products Laboratory, ** One Gifford Pinchot Drive, Madison, WI 53705-2398 ABSTRACT This research examined the titers of hexokinase (HK), phosphofruc- tokinase (PFK), and xylulokinase (XUK) in Saccharomyces cerevisiae and two xylose fermenting yeasts, Pachysolen tannophilus and Candida she- hatae, following shifts in carbon source and aeration. Xylose-grown C. shehatae, glucose-grown P. tannophilus, and glucose-grown S. cerevisiae, had the highest specific activities of XUK, HK, and PFK, respectively. XUK was induced by xylose to moderate levels in both P. tannophilus and C. shehatae, but was present only in trace levels in S. cerevisiae. HK activi- ties in P. tannophilus were two to three fold higher when cells were grown on glucose than when grown on xylose, but HK levels were less inducible in C. shehatae. The PFK activities in S. cerevisiae were 1.5 to 2 times higher than in the two xylose-fermenting yeasts. Transfer from glucose to xylose rapidly inactivated HK in P. tannophilus, and transfer from xylose to glucose inactivated XUK in C. shehatae. The patterns of induction and inactivation indicate that the basic regulatory mechanisms differ in the two xylose fermenting yeasts. Index Entries: Saccharomyces cerevisiae; Pachysolen tannophilus; Candida shehatae; 6-phosphofructokinase; hexokinase; D-xylulokinase; regulation; phosphotransferase. * Author to whom all correspondence and reprint requests should be addressed. **Maintained in cooperation with the University of Wisconsin, Madison, WI This article was written and prepared by U.S. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

Dema and Faust Et Al., Suppl. Material 2020.02.03
Supplementary Materials Cyclin-dependent kinase 18 controls trafficking of aquaporin-2 and its abundance through ubiquitin ligase STUB1, which functions as an AKAP Dema Alessandro1,2¶, Dörte Faust1¶, Katina Lazarow3, Marc Wippich3, Martin Neuenschwander3, Kerstin Zühlke1, Andrea Geelhaar1, Tamara Pallien1, Eileen Hallscheidt1, Jenny Eichhorst3, Burkhard Wiesner3, Hana Černecká1, Oliver Popp1, Philipp Mertins1, Gunnar Dittmar1, Jens Peter von Kries3, Enno Klussmann1,4* ¶These authors contributed equally to this work 1Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Robert- Rössle-Strasse 10, 13125 Berlin, Germany 2current address: University of California, San Francisco, 513 Parnassus Avenue, CA 94122 USA 3Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, 13125 Berlin, Germany 4DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Oudenarder Strasse 16, 13347 Berlin, Germany *Corresponding author Enno Klussmann Max Delbrück Center for Molecular Medicine Berlin in the Helmholtz Association (MDC) Robert-Rössle-Str. 10, 13125 Berlin Germany Tel. +49-30-9406 2596 FAX +49-30-9406 2593 E-mail: [email protected] 1 Content 1. CELL-BASED SCREENING BY AUTOMATED IMMUNOFLUORESCENCE MICROSCOPY 3 1.1 Screening plates 3 1.2 Image analysis using CellProfiler 17 1.4 Identification of siRNA affecting cell viability 18 1.7 Hits 18 2. SUPPLEMENTARY TABLE S4, FIGURES S2-S4 20 2 1. Cell-based screening by automated immunofluorescence microscopy 1.1 Screening plates Table S1. Genes targeted with the Mouse Protein Kinases siRNA sub-library. Genes are sorted by plate and well. Accessions refer to National Center for Biotechnology Information (NCBI, BLA) entries. The siRNAs were arranged on three 384-well microtitre platres. -

Microbial Cell Factories Biomed Central
Microbial Cell Factories BioMed Central Research Open Access Engineering of xylose reductase and overexpression of xylitol dehydrogenase and xylulokinase improves xylose alcoholic fermentation in the thermotolerant yeast Hansenula polymorpha Olena V Dmytruk1, Kostyantyn V Dmytruk1, Charles A Abbas2, Andriy Y Voronovsky1 and Andriy A Sibirny*1,3 Address: 1Institute of Cell Biology, NAS of Ukraine, Drahomanov Street 14/16, Lviv 79005, Ukraine, 2Archer Daniels Midland Co Research Center, 1001 N Brush College Rd., Decatur IL 62521, USA and 3Department of Microbiology and Biotechnology, Rzeszow University, Cwiklinskiej 2, 35- 601, Rzeszow, Poland Email: Olena V Dmytruk - [email protected]; Kostyantyn V Dmytruk - [email protected]; Charles A Abbas - [email protected]; Andriy Y Voronovsky - [email protected]; Andriy A Sibirny* - [email protected] * Corresponding author Published: 23 July 2008 Received: 11 June 2008 Accepted: 23 July 2008 Microbial Cell Factories 2008, 7:21 doi:10.1186/1475-2859-7-21 This article is available from: http://www.microbialcellfactories.com/content/7/1/21 © 2008 Dmytruk et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The thermotolerant methylotrophic yeast Hansenula polymorpha is capable of alcoholic fermentation of xylose at elevated temperatures (45 – 48°C). Such property of this yeast defines it as a good candidate for the development of an efficient process for simultaneous saccharification and fermentation. However, to be economically viable, the main characteristics of xylose fermentation of H. -
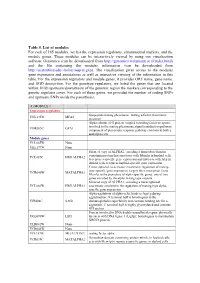
Table 5. List of Modules for Each of 165 Modules, We List the Expression Regulators, Chromosomal Markers, and the Module Genes
Table 5. List of modules For each of 165 modules, we list the expression regulators, chromosomal markers, and the module genes. These modules can be interactively viewed by using our visualization software Genomica (can be downloaded from http://genomica.weizmann.ac.il/index.html) and the file containing the modules information (can be downloaded from http://ai.stanford.edu/~koller/seqvar.gxp). The visualization gives access to the modules gene expression and annotations as well as interactive viewing of the information in this table. For the expression regulators and module genes, it provides ORF name, gene name, and SGD desciprtion. For the genotype regulators, we listed the genes that are located within 10 kb upstream/downstream of the genomic region the markers corresponding to the genetic regulator cover. For each of these genes, we provided the number of coding SNPs and upstream SNPs inside the parenthesis. (1) MODULE 1 Expression regulators lipopeptide mating pheromone; mating a-factor pheromone YNL145W MFA2 precursor Alpha subunit of G protein coupled to mating factor receptors, involved in the mating pheromone signal transduction pathway; YHR005C GPA1 component of pheromone response pathway common to both a and alpha cells Module genes YCL065W None YKL177W None Silenced copy of ALPHA2, encoding a homeobox-domain containing protein that associates with Mcm1p in haploid cells YCL067C HMLALPHA2 to repress a-specific gene expression and interacts with A1p in diploid cells to repress haploid-specific gene expression Transcriptional