BMC Structural Biology Biomed Central
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

In the Field of Clinical Examination, the Measurement of Creatine
J. Clin. Biochem. Nutr., 3, 17-25, 1987 Bacterial Glucokinase as an Enzymic Reagent of Good Stability for Measurement of Creatine Kinase Activity Hitoshi KONDO, * Takanari SHIRAISHI, Masao KAGEYAMA, Kazuhiko NAGATA, and Kosuke TOMITA Research and Development Center, UNITIKA Ltd., Uji 611, Japan (Received January 10, 1987) Summary An enzymic reagent, that has long-term stability even in the liquid state, was successfully employed for the measurement of serum creatine kinase (CK, EC 2.7.3.2) activity. The enzyme used was the thermostable glucokinase (GlcK, EC 2.7.1.2) obtained from the thermo- phile Bacillus stearothermophilus. The reagent was found to be stable in solution for about one month at 6•Ž and for about one week at 30•Ž. This substitution of glucokinase for the hexokinase of the most commonly used hexokinase-glucose-6-phosphate dehydrogenase (HK-G6PDH) method results in a remarkable improvement of the method. The CK activity measured by the GlcK-G6PDH method was linear up to about 2,000 U/ liter at 37•Ž. The GlcK-G6PDH method was found to give a satisfactory precision and reproducibility (coefficient of variation less than 2.17%). Over a wide range of CK activity, an excellent agreement was obtained between the GlcK-G6PDH and the HK-G6PDH methods. Furthermore several coexistents and anticoagulants were found to have little effect on the measured value of CK activity by the GlcK-G6PDH method. Key Words: creatine kinase activity, glucokinase, improved stability of reagent, creatine kinase determination, thermostable enzyme In the field of clinical examination, the measurement of creatine kinase (CK, ATP : creatine phosphotransferase, EC 2.7.3.2) activity in serum is one of the important examinations usually employed for diagnosis of cardiac diseases such as myocardial infarction or muscular diseases such as progressive muscular dystrophy. -

Creatine Kinase Assay Kit
Creatine Kinase Assay Kit Catalog Number KA1665 100 assays Version: 03 Intended for research use only www.abnova.com Table of Contents Introduction ................................................................................................... 3 Intended Use ................................................................................................................. 3 Background ................................................................................................................... 3 Principle of the Assay .................................................................................................... 3 General Information ...................................................................................... 4 Materials Supplied ......................................................................................................... 4 Storage Instruction ........................................................................................................ 4 Materials Required but Not Supplied ............................................................................. 4 Precautions for Use ....................................................................................................... 4 Assay Protocol .............................................................................................. 5 Reagent Preparation ..................................................................................................... 5 Sample Preparation ...................................................................................................... -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Polymerase Ribozyme with Promoter Recognition
In vitro Evolution of a Processive Clamping RNA Polymerase Ribozyme with Promoter Recognition by Razvan Cojocaru BSc, Simon Fraser University, 2014 Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Molecular Biology and Biochemistry Faculty of Science © Razvan Cojocaru 2021 SIMON FRASER UNIVERSITY Summer 2021 Copyright in this work is held by the author. Please ensure that any reproduction or re-use is done in accordance with the relevant national copyright legislation. Declaration of Committee Name: Razvan Cojocaru Degree: Doctor of Philosophy Title: In vitro Evolution of a Processive Clamping RNA Polymerase Ribozyme with Promoter Recognition Committee: Chair: Lisa Craig Professor, Molecular Biology and Biochemistry Peter Unrau Supervisor Professor, Molecular Biology and Biochemistry Dipankar Sen Committee Member Professor, Molecular Biology and Biochemistry Michel Leroux Committee Member Professor, Molecular Biology and Biochemistry Mani Larijani Internal Examiner Associate Professor, Molecular Biology and Biochemistry Gerald Joyce External Examiner Professor, Jack H. Skirball Center for Chemical Biology and Proteomics Salk Institute for Biological Studies Date Defended/Approved: August 12, 2021 ii Abstract The RNA World hypothesis proposes that the early evolution of life began with RNAs that can serve both as carriers of genetic information and as catalysts. Later in evolution, these functions were gradually replaced by DNA and enzymatic proteins in cellular biology. I start by reviewing the naturally occurring catalytic RNAs, ribozymes, as they play many important roles in biology today. These ribozymes are central to protein synthesis and the regulation of gene expression, creating a landscape that strongly supports an early RNA World. -
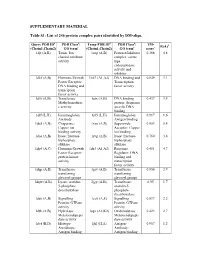
List of 246 Protein Complex Pairs Identified by DM-Align
SUPPLEMENTARY MATERIAL Table S1: List of 246 protein complex pairs identified by DM-align. Query PDB IDa PDB Classb: Temp PDB IDd PDB Classb: TM- R(Å)f (Chain1,Chain2) GO termc (Chain1,Chain2) GO termc scoree 1djt (A,B) Toxin: Ion 1aap (A,B) Protease/Inhibitor 0.368 4.8 channel inhibitor complex: serine activity type endopeptidase activity and inhibitor 1dkf (A,B) Hormone/Growth 1xb7 (A1,A2) DNA binding and 0.849 3.1 Factor Receptor: Transcription DNA binding and factor activity transciption factor activity 1dl5 (A,B) Transferase: 1utx (A,B) DNA binding 0.437 3.9 Methyltransferas protein: Sequence e activity specific DNA binding 1dlf (L,H) Immunoglobin: 1j05 (L,H) Immunoglobin: 0.917 1.6 Antibody Antigen binding 1do5 (A,B) Chaperone: 1xso (A,B) Superoxide 0.953 0.9 Copper ion Acceptor: Copper binding activity ion binding 1dos (A,B) lyase: fructose- 1rvg (A,B) lyase: fructose- 0.760 3.6 biphosphate biphosphate aldolase aldolase 1dp4 (A,C) Hormone/Growth 1dz3 (A1,A2) Response 0.481 4.7 Factor Receptor: Regulator: DNA protein kinase binding and activity transcription factor activity 1dqp (A,B) Transferase: 1grv (A,B) Transferase: 0.856 2.9 transferring transferring glycosyl groups glycosyl groups 1dqw (A,B) Lyase: orotidin- 2jgy (A,B) Transferase: 0.95 1.7 5-phosphate orotidin-5- decarboxylase phosphate decarboxylase 1ds6 (A,B) Signalling 1cc0 (A,E) Signalling 0.897 2.2 Protein: GTPase Protein: GTPase activity activity 1dth (A,B) Hydrolase: 1sqv (A2,K2) Oxidoredutase: 0.423 2.7 Metaloendopepti Metaloendopepti dase activity dase activity 1dvf -

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Ceramide Metabolism Regulates a Neuronal Nadph Oxidase Influencing Neuron Survival During Inflammation
Ceramide Metabolism Regulates A Neuronal Nadph Oxidase Influencing Neuron Survival During Inflammation Item Type Thesis Authors Barth, Brian M. Download date 07/10/2021 02:29:56 Link to Item http://hdl.handle.net/11122/8999 CERAMIDE METABOLISM REGULATES A NEURONAL NADPH OXIDASE INFLUENCING NEURON SURVIVAL DURING INFLAMMATION A THESIS Presented to the Faculty of the University of Alaska Fairbanks in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY By Brian M. Barth, B.S., M.S. Fairbanks, Alaska August 2009 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. UMI Number: 3386045 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI Dissertation Publishing UMI 3386045 Copyright 2009 by ProQuest LLC. All rights reserved. This edition of the work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. CERAMIDE METABOLISM REGULATES A NEURONAL NADPH OXIDASE INFLUENCING NEURON SURVIVAL DURING INFLAMMATION By Brian M. Barth RECOMMENDED: , ... f / M Advisory Committee Chah Chai ^Department of Chemistry and Biochemistry /? & f ) ./ APPROVED: Dean, C olleg£^ Natural Science'and“Mathemati cs /<C £>dan of the Graduate School 3 / / - Date Reproduced with permission of the copyright owner. -

When a Slight Tilt Is Enough
Cell. Mol. Life Sci. (2013) 70:181–203 DOI 10.1007/s00018-012-1038-x Cellular and Molecular Life Sciences REVIEW Ceramide function in the brain: when a slight tilt is enough Chiara Mencarelli • Pilar Martinez–Martinez Received: 11 January 2012 / Revised: 16 May 2012 / Accepted: 21 May 2012 / Published online: 24 June 2012 Ó The Author(s) 2012. This article is published with open access at Springerlink.com Abstract Ceramide, the precursor of all complex sphin- Introduction golipids, is a potent signaling molecule that mediates key events of cellular pathophysiology. In the nervous system, Ceramides are a family of lipid molecules that consist of the sphingolipid metabolism has an important impact. sphingoid long-chain base linked to an acyl chain via an Neurons are polarized cells and their normal functions, amide bond. Ceramides differ from each other by length, such as neuronal connectivity and synaptic transmission, hydroxylation, and saturation of both the sphingoid base rely on selective trafficking of molecules across plasma and fatty acid moieties. membrane. Sphingolipids are abundant on neural cellular Sphingoid bases are of three general chemical types: membranes and represent potent regulators of brain sphingosine, dihydrosphingosine (commonly known as homeostasis. Ceramide intracellular levels are fine-tuned ‘‘sphinganine’’, as it will be addressed in this review) and and alteration of the sphingolipid–ceramide profile con- phytosphingosine. Based on the nature of the sphingoid tributes to the development of age-related, neurological base backbone, we can distinguish three main subgroups in and neuroinflammatory diseases. The purpose of this the ceramide family: the compound named ceramide con- review is to guide the reader towards a better understanding tains sphingosine, which has a trans-double bond at the of the sphingolipid–ceramide pathway system. -

HOXB6 Homeo Box B6 HOXB5 Homeo Box B5 WNT5A Wingless-Type
5 6 6 5 . 4 2 1 1 1 2 4 6 4 3 2 9 9 7 0 5 7 5 8 6 4 0 8 2 3 1 8 3 7 1 0 0 4 0 2 5 0 8 7 5 4 1 1 0 3 6 0 4 8 3 7 4 7 6 9 6 7 1 5 0 8 1 4 1 1 7 1 0 0 4 2 0 8 1 1 1 2 5 3 5 0 7 2 6 9 1 2 1 8 3 5 2 9 8 0 6 0 9 5 1 9 9 2 1 1 6 0 2 3 0 3 6 9 1 6 5 5 7 1 1 2 1 1 7 5 4 6 6 4 1 1 2 8 4 7 1 6 2 7 7 5 4 3 2 4 3 6 9 4 1 7 1 3 4 1 2 1 3 1 1 4 7 3 1 1 1 1 5 3 2 6 1 5 1 3 5 4 5 2 3 1 1 6 1 7 3 2 5 4 3 1 6 1 5 3 1 7 6 5 1 1 1 4 6 1 6 2 7 2 1 2 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S HOXB6 homeo box B6 HOXB5 homeo box B5 WNT5A wingless-type MMTV integration site family, member 5A WNT5A wingless-type MMTV integration site family, member 5A FKBP11 FK506 binding protein 11, 19 kDa EPOR erythropoietin receptor SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 RAD52 RAD52 homolog S.