Handbook on the Physics and Chemistry of Rare Earths Volume 9 Elsevier, 1987
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Standard X-Ray Diffraction Powder Patterns
NBS MONOGRAPH 25 — SECTION 1 Standard X-ray Diffraction U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS Functions and Activities The functions of the National Bureau of Standards are set forth in the Act of Congress, March 3, 1901, as amended by Congress in Public Law 619, 1950. These include the development and maintenance of the national standards of measurement and the provision of means and methods for making measurements consistent with these standards; the determination of physical constants and properties of materials; the development of methods and instruments for testing materials, devices, and structures; advisory services to government agencies on scien- tific and technical problems; invention and development of devices to serve special needs of the Government; and the development of standard practices, codes, and specifications. The work includes basic and applied research, development, engineering, instrumentation, testing, evaluation, calibration services, and various consultation and information services. Research projects are also performed for other government agencies when the work relates to and supplements the basic program of the Bureau or when the Bureau's unique competence is required. The scope of activities is suggested by the listing of divisions and sections on the inside of the back cover. Publications The results of the Bureau's research are published either in the Bureau's own series of publications or in the journals of professional and scientific societies. The Bureau itself publishes three periodicals available from the Government Printing Office: The Journal of Research, published in four separate sections, presents complete scientific and technical papers; the Technical News Bulletin presents summary and preliminary reports on work in progress; and Basic Radio Propagation Predictions provides data for determining the best frequencies to use for radio communications throughout the world. -
Evaluation of Aqueous Systems
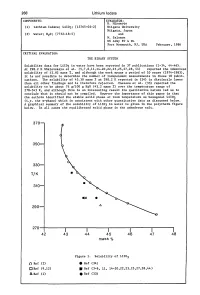
268 Lithium Iodate COMPONENTS: EVALUATOR: H. Miyamoto (1) Lithium Iodate; LiI03; [13765-03-2] Niigata University Niigata, Japan (2) Water; H20; [7732-18-5] and M. Salomon US Army ET & DL Fort Monmouth, NJ, USA February, 1986 CRITICAL EVALUATION: THE BINARY SYSTEM Solubility data for LiI03 in water have been reported in 37 publications (1-34, 44-46). At 298.2 K Shklovskaya et al. (5,7,8,11,14-20,22,23,25,27,28,44) reported the identical solubility of 43.82 mass %, and although the work spans a period of 10 years (1974-1983), it is not possible to determine the number of independent measurements in these 18 publi cations. The solubility of 43.30 mass % at 298.2 K reported in (24) is distinctly lower than all other findings and is therefore rejected. Unezawa et al. (33) reported the solubility to be about 76 g/lOO g H20 (43.2 mass %) over the temperature range of 278-343 K, and although this is an interesting result its qualitative nature led us to conclude that it should not be compiled. However the importance of this paper is that the authors identified the stable solid phase at room temperature as hexagonal LiI03 (i.e. the a-phase) which is consistent with other quantitative data as discussed below. A graphical summary of the solubility of LiI03 in water is given in the polytherm figure below. In all cases the equilibrated solid phase is the anhydrous salt. 370 350 330 T/K 310 290 270-I------.r-----.-----..----......----------. 42 43 44 45 46 47 48 mass % Figure 1. -
![PDF Card 3-320 [Dow Chemical Co., Midland, 1 .3805 1 006 67.83 Michigan] 1 .3672 6 300 68.58](https://docslib.b-cdn.net/cover/4533/pdf-card-3-320-dow-chemical-co-midland-1-3805-1-006-67-83-michigan-1-3672-6-300-68-58-1214533.webp)
PDF Card 3-320 [Dow Chemical Co., Midland, 1 .3805 1 006 67.83 Michigan] 1 .3672 6 300 68.58
standard X-ray Diffraction Powder Patterns ^v^iSection 10-Data for 84 Substances ^•2. — Howard E. Swanson, Howard F. McMurdie, Marlene C. Morris lliloise H. Evans, and Boris Paretzkin Assisted by Johan H. deGroot and Simon J. Carmel Institute for Materials Research -y.J National Bureau of Standards ' Washington, D.C. 20234 U.S. DEPARTMENT OF COMMERCE, Refer G. Peterson, Secretary NATIONAL BUREAU OF STANDARDS, Lawrence M. Kushner, AcUng Director, Issued November 1972 Library of Congress Catalog Card Number: 53—61386 National Bureau of Standards Monograph 25 Section 10—Data for 84 Substances Nat. Bur. Stand. (U.S.), Monogr. 25— Sec. 10,161 pages (Nov. 1972) CODEN: NBSMA6 For sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, D.C. 20402 (Order by SD Catalog No. C13.44: 25/Sec. 10). Price $2.00 CONTENTS Page Page Introduction 1 Zinc manganese oxide (hetaerolite), ZnMn20^ 61 Experimental patterns: Zinc tin oxide, Zn2Sn04 62 Ammonium aluminum sulfate, NH^AKSO^)^ 5 Calculated patterns: Ammonium copper bromide hydrate, (NH^)2CuBr^"2H20 .. 6 Barium bromide, BaBr2 63 Ammonium iodate, NH^IOj 7 Barium iodide, Bal2 66 Ammonium iron sulfate, NH^Fe(S0^)2 8 Boron oxide, B2O3 phase 1 70 Ammonium magnesium aluminum fluoride, NH^IVIgAIFg ... 9 Calcium iron silicate hydroxide, julgoldite, Barium bromide fluoride, BaBrF 10 Ca2Fe3Si30jo(OH,0)2(OH)2 72 Barium chloride fluoride, BaCIF 11 Calcium malate hydrate, CaC4H405-2H20 76 Barium sulfate (barite), BaSO^ (revised). 12 Cesium lithium cobalt cyanide, CsLiCo(CN)g 79 Cadmium -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Studies on the Growth and Characterization of Some Optical Crystals
STUDIES ON THE GROWTH AND CHARACTERIZATION OF SOME OPTICAL CRYSTALS Thesis of the research work submitted to Bharathidasan University, Thiruchirappalli in partial fulfillment of the requirements for the award of the degree of DOCTOR OF PHILOSOPHY IN PHYSICS Submitted by P.PARAMASIVAM Under the Supervision of Dr. C. RAMACHANDRA RAJA, Ph.D., Associate Professor in Physics POSTGRADUATE & RESEARCH DEPARTMENT OF PHYSICS GOVERNMENT ARTS COLLEGE (AUTONOMOUS) KUMBAKONAM - 612001 TAMIL NADU, INDIA FEBRUARY – 2012 Dr. C. Ramachandra Raja, Ph.D., Associate Professor in Physics, Department of Physics, Government Arts College (Autonomous), Phone : +91 4364221751 Kumbakonam – 612001, CelL: +91 9976696277 Tamil Nadu, India. Email: [email protected] CERTIFICATE This is to certify that the thesis entitled “STUDIES ON THE GROWTH AND CHARACTERIZATION OF SOME OPTICAL CRYSTALS” submitted by Mr. P.PARAMASIVAM is a bonafide record of the research work done by him during the period of study from 2004 to 2011 under my supervision in the Department of Physics, Government Arts College (Autonomous), Kumbakonam and that the thesis has not previously formed the basis for the award of any Degree, Diploma, Associateship, Fellowship or any other similar title. This thesis represents an independent work on the part of candidate. Kumbakonam C. Ramachandra Raja (Research Supervisor) Mr. P.Paramasivam, Research Scholar (Part - Time), Department of Physics, Government Arts College (Autonomous), Kumbakonam – 612001, Tamil Nadu, India. DECLARATION I hereby declare that the work presented in this thesis entitled “STUDIES ON THE GROWTH AND CHARACTERISATION OF SOME OPTICAL CRYSTALS” has been originally carried out by me under the guidance and supervision of Dr.C.Ramachandra Raja, Associate Professor, Department of Physics, Government Arts College (Autonomous), Kumbakonam. -

Ep 0083096 B1
Patentamt JEuropâisches European Patent Office ® Publication number: Q Q83 096 Office européen des brevets g "jj © EUROPEAN PATENT SPECIFICATION (45) Dateof publication of patent spécification: 25.02.87 (g) Int. Cl.4: C 07 C 125/06 ® Application number: 82111989.8 @ Date offiling: 24.12.82 (!) Additional priorities: 26.02.82 JP 2887582; 01.03.82 JP 3057982; 03.03.82 JP 3221382; 15.03.82 JP 3934982; 15.03.82 JP 3935082; 24.03.82 JP 4566782; 26.03.82 JP 4832482. (H) Production of urethane compounds. (§) Priority: 25.12.81 JP 213191/81 (73) Proprietor: Asahi Kasei Kogyo Kabushiki Kaisha 30.12.81 JP 210640/81 2-6, Dojimahama 1 -chôme Kita-ku 19.01.82 JP 5356/82 Osaka-shi Osaka 530 (JP) 19.01.82 JP 5357/82 25.01.82 JP 8840/82 25.01 .82 JP 8841/82 (72) Inventor: Fukuoka, Shinsuke 28.01 .82 JP 10862/82 Asahi-Kasei-Otaka-Apt. 2-904 1005-1, 28.01.82 JP 10863/82 Higashitomii 28.01. 82 JP 10864/82 Kurashiki-shi Okayama-ken (JP) 28.01. 82 JP 10865/82 Inventor: Chono, Masazumi 05.02.82 JP 16442/82 151-3, Yasue 22.02.82 JP 26144/82 Kurashiki-shi Oakayama-ken (JP) 22.02.82 JP 26145/82 23.02.82 JP 26750/82 24.02.82 JP 27290/82 (7Ï) Représentative: Boeters, Hans Dietrich, Dr. et al 24.02.82 JP 28532/82 Boeters, Bauer & Partner Thomas-Wimmer-Ring 25.02.82 JP 28106/82 14 25.02.82 JP 28107/82 D-8000 Mûnchen 22 (DE) 25.02.82 JP 29170/82 3T 26.02.82 JP 28874/82 OÙ (43) Date of publication of application: (84) Designated Contracting States: (0 06.07.83 Bulletin 83/27 BE DE FR GB IT NL O) (§) Références cited: O ® Publication ofthegrantofthe patent: 25.02.87 Bulletin 87/09 EP-A-0 016346 EP-A-0 036 895 00 o Note: Within nme months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European Notice of shall CL patent granted. -
Ternary Systems
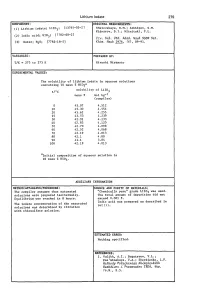
Lithium Iodate 279 COMPONENTS: ORIGINAL MEASUREMENTS: Shklovskaya, R.M.; Arkhipov, S.M. (1) Lithium iodate; Li103; [13765-03-2] Kidyarov, B.l.; Mitnitski, P.L. (2) lodic acid; HI03; [7782-68-2] lzv. S~b. Otd. AQad. NauQ SSSR S~. (3) Water; H20; [7782-18-5] Khim. NauQ 1976, (61, 89-91. VARIABLES: PREPARED BY: T/K = 273 to 373 K Hiroshi Miyamoto EXPERIMENTAL VALUES: The solubility of lithium iodate in aqueous solutions containing 10 mass % Hl03a t/OC solubility of Lil03 mass % mol kg-l (compiler) o 45.07 4.512 10 45.30 4.554 20 43.62 4.255 25 43.53 4.239 30 42.91 4.133 40 42.83 4.120 50 42.70 4.098 60 42.52 4.068 70 42.19 4.013 80 42.1 4.00 90 41.1 3.84 100 42.19 4.013 aInitial composition of aqueous solution is 10 mass % HI03 . AUXILIARY INFORMATION METHOD/APPARATUS/PROCEDURE: SOURCE AND PURITY OF MATERIALS: The compiler assumes that saturated "Chemically pure" grade Lil03 was used. solutions were prepared isothermally. The total amount of impurities did not Equilibrium was reached in 8 hours. exceed 0.001 %. lodic acid was prepared as described in The iodate concentration of the saturated ref (1). solutions was determined by titration with thiosulfate solution. ESTIMATED ERROR: Nothing specified. REFERENCES: 1. Vulikh, A.I.; Bogatyrev, V.L.; Kaz'minskaya, V.A.; Zherdienko, L.P. Me.thodq Po.euche.u.qa KhimcheAUQh Reallivov ~ PltepaJtatov lREA, VCfP. 16.M., 5.5. 280 Lithium Iodate COMPONENTS: ORIGINAL HEASUREHENTS: (1) Lithium iodate; LiI03; [13765-03-2] Tarasova, G.N.; Vinogradov, E.E.; Lepeshkov, I.N. -

Elasto-Optic, Electro-Optic, and Magneto-Optic Constants
ELASTO-OptIC, ELECTRO-OptIC, AND MAGNETO-OptIC CONSTANTS When a crystal is subjected to a stress field, an electric field, or The coefficients rij,k have the name (linear) electro-optic coef- a magnetic field, the resulting optical effects are in general depen- ficients . dent on the orientation of these fields with respect to the crys- The values of the electro-optic coefficients depend on the tal axes . It is useful, therefore, to express the optical properties in boundary conditions . If the superscripts T and S denote, respec- terms of the refractive index ellipsoid (or indicatrix): tively, the conditions of zero stress (free) and zero strain (clamped) 2 2 2 one finds: x ++y z = 2 2 2 1 TS=+ES=+E nxyn nz rrij ij qeik jk rPij ik d jk or where ejk = (∂Tk/∂Ej)S and djk = (∂Sk/∂Ej)T are the appropriate piezo- electric coefficients . == ∑ Bxij ijyi11(,j ,,)23 The interaction between a magnetic field and a light wave prop- ij agating in a solid or in a liquid gives rise to a rotation of the plane of polarization . This effect is known as Faraday rotation . It results where from a difference in propagation velocity for left and right circular polarized light . 11 B = = The Faraday rotation, θ , is linearly proportional to the mag- ij ε 2 F ij n ij netic field H: θ = F VlH ε is the dielectric constant or permeability; the quantity Bij is called impermeability . A crystal exposed to a stress S will show a change of its imper- where l is the light path length and V is the Verdet constant (min- utes/oersted·cm) . -

Tilley R.J.D. Colour and the Optical Properties of Materials (Wiley, 2011
Colour and the Optical Properties of Materials An Exploration of the Relationship Between Light, the Optical Properties of Materials and Colour PROFESSOR RICHARD J. D. TILLEY Emeritus Professor, University of Cardiff, UK Colour and the Optical Properties of Materials Colour and the Optical Properties of Materials An Exploration of the Relationship Between Light, the Optical Properties of Materials and Colour PROFESSOR RICHARD J. D. TILLEY Emeritus Professor, University of Cardiff, UK This edition first published 2011 Ó 2011 John Wiley & Sons, Ltd Registered office John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, United Kingdom For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com. The right of the author to be identified as the author of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, withoutthe prior permission of the publisher. Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. -
Database Full Listing
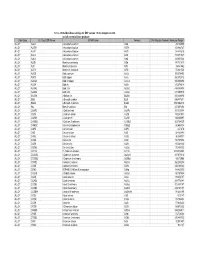
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -
Standard X-Ray Diffraction Powder Patterns
E^l Admin. NBS MONOGRAPH 25—SECTION 5 Refecii^M not to be ^ferlrom the library. Standard X-ray Diffraction Powder Patterns ^\ / U.S. DEPARTMENT OF COMMERCE S NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS The National Bureau of Standards^ provides measurement and technical information services essential to the efficiency and effectiveness of the work of the Nation's scientists and engineers. The Bureau serves also as a focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. To accomplish this mission, the Bureau is organized into three institutes covering broad program areas of research and services: THE INSTITUTE FOR BASIC STANDARDS . provides the central basis within the United States for a complete and consistent system of physical measurements, coordinates that system with the measurement systems of other nations, and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. This Institute comprises a series of divisions, each serving a classical subject matter area: —Applied Mathematics—Electricity—Metrology—Mechanics—Heat—Atomic Physics—Physical Chemistry—Radiation Physics— -Laboratory Astrophysics^—Radio Standards Laboratory,^ which includes Radio Standards Physics and Radio Standards Engineering—Office of Standard Refer- ence Data. THE INSTITUTE FOR MATERIALS RESEARCH . conducts materials research and provides associated materials services including mainly reference materials and data on the properties of ma- terials. Beyond its direct interest to the Nation's scientists and engineers, this Institute yields services which are essential to the advancement of technology in industry and commerce. -

Standard X-Ray Diffraction Powder Patterns
7 NATL INST OF STANDARDS & TECH R.I.C. Nes AlllOD Ififib^fi PUBLICATIONS 100988698 ST5°5rv'25-17;1980 C.1 NBS-PUB-C 19 cr»T OF NBS MONOGRAPH 25-SECTION 1 V) J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards Standard X-ray Diffraction Powder Patterns . NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act ot Congress on March 3, 1901 The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau's technical work is per- formed by the National Measurement Laboratory, the National Engineering Laboratory, and the Institute for Computer Sciences and Technology. THE NATIONAL MEASUREMENT LABORATORY provides the national system of physical and chemical and materials measurement; coordinates the system with measurement systems of other nations and furnishes essential services leading to accurate and uniform physical and chemical measurement throughout the Nation's scientific community, industry, and commerce; conducts materials research leading to improved methods ol measurement, standards, and data on the properties of materials needed by industry, commerce, educational institutions, and Government; provides advisory and research services to other Government agencies; develops, produces, and distributes Standard Reference Materials; and provides calibration services. The Laboratory consists of the following centers: Absolute Physical Quantities- — Radiation Research — Thermodynamics and Molecular Science — Analytical Chemistry — Materials Science.