Discovering Conglomerates for Chiral Resolution by Crystallization Aliou Mbodji
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
A Practical Approach to Chiral Separations by Liquid Chromatography
A Practical Approach to Chiral Separations by Liquid Chromatography Edited by G. Subramanian Weinheim • New York VCH Basel • Cambridge • Tokyo Contents 1 An Introduction to Enantioseparation by Liquid Chromatography Charles A. White and Ganapathy Subramanian 1.1 Introduction 1 1.1.1 What is Chirality? 1 1.1.2 What Causes Chirality? 2 1.1.3 Why is Chirality Important? 4 1.2 Industries which Require Enantioseparations 4 1.2.1 Pharmaceutical Industry 4 1.2.2 Agrochemical Industry 5 1.2.3 Food and Drink Industry 6 1.2.4 Petrochemical Industry 6 1.3 Chiral Liquid Chromatography 7 1.3.1 Type I Chiral Phases 8 1.3.2 Type II Chiral Stationary Phases 10 1.3.3 Type III Chiral Stationary Phases 10 1.3.3.1 Microcrystalline Cellulose Triacetate and Tribenzoate 10 1.3.3.2 Cyclodextrins 11 1.3.3.3 Crown Ethers 11 1.3.3.4 Synthetic Polymers •. 12 1.3.4 Type IV Chiral Stationary Phases 13 1.3.5 Type V Chiral Stationary Phases 13 1.3.6 Mobile Phase Additives 14 1.3.6.1 Metal Complexes 14 1.3.6.2 Ion-pair Formation 15 1.3.6.3 Uncharged Chiral Additives 15 1.4 The Future 16 2 Modeling Enantiodifferentiation in Chiral Chromatography Kenny B. Lipkowitz 2.1 Introduction 19 2.2 Modeling 19 # VIII Contents 2.3 Computational Tools 22 2.3.1 Quantum Mechanics 22 2.3.2 Molecular Mechanics 23 2.3.3 Molecular Dynamics 24 2.3.4 Monte Carlo Simulations 24 2.3.5 Graphics 25 2.4 Modeling Enantioselective Binding in Chromatography 26 2.4.1 Type I CSPs 26* 2.4.2 Type II CSPs 41 2.4.3 Type III CSPs 44 2.5 Related Studies 49 2.6 Summary 50 3 Regulatory Implications and Chiral Separations Jeffrey R. -

Standard X-Ray Diffraction Powder Patterns
NBS MONOGRAPH 25 — SECTION 1 Standard X-ray Diffraction U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS Functions and Activities The functions of the National Bureau of Standards are set forth in the Act of Congress, March 3, 1901, as amended by Congress in Public Law 619, 1950. These include the development and maintenance of the national standards of measurement and the provision of means and methods for making measurements consistent with these standards; the determination of physical constants and properties of materials; the development of methods and instruments for testing materials, devices, and structures; advisory services to government agencies on scien- tific and technical problems; invention and development of devices to serve special needs of the Government; and the development of standard practices, codes, and specifications. The work includes basic and applied research, development, engineering, instrumentation, testing, evaluation, calibration services, and various consultation and information services. Research projects are also performed for other government agencies when the work relates to and supplements the basic program of the Bureau or when the Bureau's unique competence is required. The scope of activities is suggested by the listing of divisions and sections on the inside of the back cover. Publications The results of the Bureau's research are published either in the Bureau's own series of publications or in the journals of professional and scientific societies. The Bureau itself publishes three periodicals available from the Government Printing Office: The Journal of Research, published in four separate sections, presents complete scientific and technical papers; the Technical News Bulletin presents summary and preliminary reports on work in progress; and Basic Radio Propagation Predictions provides data for determining the best frequencies to use for radio communications throughout the world. -

Chiral Resolution Screening and Purification Kits Brochure
Maybridge Chiral Resolution Screening and Purification Kits Offering rapid access to optically pure chiral compounds Maybridge Chiral Resolution Screening and Purification Kits Introduction Diastereomeric crystallization is a commonly used effective process to obtain optically pure chiral compounds from their racemic mixtures. However, choosing the optimal conditions for the process; e.g., combination of resolving agents and solvents, is time-consuming, tedious and labor-intensive. Maybridge Chiral Resolution Screening and Purification kits provide scientists with a quick and systematic approach to find the best separation conditions under which the target compound can be isolated with the highest yield and optical purity. Key features and benefits • Rapid Screening – the kits include 384 different combinations of resolving agents and solvents, increasing the chances of finding the optimal separation conditions • High Performance – development time reduced to one day • Efficient – as little as 0.4mmol of racemate required • Ready to Use – resolving agents and solvents are pre-dispensed in 96-well plates • Convenient – the screening kits provide positive results identifiable by a quick visual or optical inspection, and the purification and recovery kit allows easy recovery and purification of the enantiomers Types of Chiral Resolution Screening and Purification Kits Amount Plate Product name Description Selection guide racemate Product code type required Maybridge Chiral • 4 x 96 plates containing 32 different acidic Identifies optimal -

A Reminder… Chirality: a Type of Stereoisomerism
A Reminder… Same molecular formula, isomers but not identical. constitutional isomers stereoisomers Different in the way their Same connectivity, but different atoms are connected. spatial arrangement. and trans-2-butene cis-2-butene are stereoisomers. Chirality: A Type of Stereoisomerism Any object that cannot be superimposed on its mirror image is chiral. Any object that can be superimposed on its mirror image is achiral. Chirality: A Type of Stereoisomerism Molecules can also be chiral or achiral. How do we know which? Example #1: Is this molecule chiral? 1. If a molecule can be superimposed on its mirror image, it is achiral. achiral. Mirror Plane of Symmetry = Achiral Example #1: Is this molecule chiral? 2. If you can find a mirror plane of symmetry in the molecule, in any achiral. conformation, it is achiral. Can subject unstable conformations to this test. ≡ achiral. Finding Chirality in Molecules Example #2: Is this molecule chiral? 1. If a molecule cannot be superimposed on its mirror image, it is chiral. chiral. The mirror image of a chiral molecule is called its enantiomer. Finding Chirality in Molecules Example #2: Is this molecule chiral? 2. If you cannot find a mirror plane of symmetry in the molecule, in any conformation, it is chiral. chiral. (Or maybe you haven’t looked hard enough.) Pharmacology of Enantiomers (+)-esomeprazole (-)-esomeprazole proton pump inhibitor inactive Prilosec: Mixture of both enantiomers. Patent to AstraZeneca expired 2002. Nexium: (+) enantiomer only. Process patent coverage to 2007. More examples at http://z.umn.edu/2301drugs. (+)-ibuprofen (-)-ibuprofen (+)-carvone (-)-carvone analgesic inactive (but is converted to spearmint oil caraway oil + enantiomer by an enzyme) Each enantiomer is recognized Advil (Wyeth) is a mixture of both enantiomers. -

Pyrethroid Stereoisomerism: Diastereomeric and Enantiomeric Selectivity in Environmental Matrices – a Review
Orbital: The Electronic Journal of Chemistry journal homepage: www.orbital.ufms.br e-ISSN 1984-6428 | Vol 10 | | No. 4 | | Special Issue June 2018 | REVIEW Pyrethroid Stereoisomerism: Diastereomeric and Enantiomeric Selectivity in Environmental Matrices – A Review Cláudio Ernesto Taveira Parente*, Claudio Eduardo Azevedo-Silva, Rodrigo Ornellas Meire, and Olaf Malm Laboratório de Radioisótopos, Instituto de Biofísica, Universidade Federal do Rio de Janeiro. Av. Carlos Chagas Filho s/n, bloco G, sala 60, subsolo - 21941-902. Cidade Universitária, Rio de Janeiro, RJ. Brasil. Article history: Received: 30 July 2017; revised: 10 October 2017; accepted: 08 March 2018. Available online: 11 March 2018. DOI: http://dx.doi.org/10.17807/orbital.v10i4.1057 Abstract: Pyrethroids are chiral insecticides characterized by stereoisomerism, which occurs due to the presence of one to three asymmetric (chiral) carbons, generating one or two pairs of cis/trans diastereomers, and two or four pairs of enantiomers. Diastereomers have equal chemical properties and different physical properties, while enantiomer pairs, have the same physicochemical properties, with exception by the ability to deviate the plane of polarized light to the right or to the left. However, stereoisomers exhibit different toxicities at the metabolic level, presenting enzyme and receptor selectivity in biological systems. Studies in aquatic organisms have shown that toxic effects are caused by specific enantiomers (1R-cis and 1R-trans) and that those cause potential estrogenic effects (1S-cis and 1S- trans). In this context, the same compound can have wide-ranging effects in organisms. Therefore, several studies have highlighted pyrethroid stereochemical selectivity in different environmental matrices. The analytical ability to distinguish the diastereomeric and enantiomeric patterns of these compounds is fundamental for understanding the processes of biotransformation, degradation environmental behavior and ecotoxicological impacts. -
![Chapter 5: Stereoisomerism [Sections: 5.1-5.9]](https://docslib.b-cdn.net/cover/1578/chapter-5-stereoisomerism-sections-5-1-5-9-331578.webp)
Chapter 5: Stereoisomerism [Sections: 5.1-5.9]
Chapter 5: Stereoisomerism [Sections: 5.1-5.9] 1. Identifying Types of Isomers Same MolecularFormula? NO YES A B C compounds are not isomers Same Connectivity? NO YES D E Different Orientation constitutional of Substituents isomers in Space? verify by: A vs B? • have different names (parent name is different or numbering [locants] of substituents) A vs C? • nonsuperimposable C vs D? YES NO D vs E? C vs E? B vs E? stereoisomers same molecule verify by: • both must have the same name • two must be superimposable P: 5.57 2. Defining Chirality • a chiral object is any object with a non-superimposable mirror image • the mirror image of a chiral object is not identical (i.e., not superimposable) • an achiral object is any object with a superimposable mirror image • the mirror image of an achiral object is identical (i.e., superimposable) # different types of groups and/or atoms the central atom is attached to: mirror image superimposable? chiral? • molecules can also be chiral • the mirror image of a chiral molecule is non-superimposable and is therefore an isomer • since the two mirror image compounds have the same connectivity, they are NOT constitutional isomers • the two mirror image compounds are nonsuperimposable due to different orientations of substituents in space: stereoisomers • non-superimposable stereoisomers that bear a mirror-image relationship are enantiomers • stereoisomers that do NOT bear a mirror-image relationship are diastereomers stereoisomers Mirror Image Relationship? NO YES diastereomers enantiomers verify by: verify by: • names differ by cis/trans, E/Z or • names differ by by having different R and S having exactly configurations at one or more opposite R and S chirality centers (but not exactly configurations at opposite configurations from each every chirality center other) 3. -

Cross-Linked Protein Crystal Technology in Bioseparation and Biocatalytic Applications
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Aaltodoc Helsinki University of Technology, Department of Chemical Technology Technical Biochemistry Report 1/2004 Espoo 2004 TKK-BE-8 CROSS-LINKED PROTEIN CRYSTAL TECHNOLOGY IN BIOSEPARATION AND BIOCATALYTIC APPLICATIONS Antti Vuolanto Dissertation for the degree of Doctor in Science in Technology to be presented with due permission of the Department of Chemical Technology for public examination and debate in Auditorium KE 2 (Komppa Auditorium) at Helsinki University of Technology (Espoo, Finland) on the 20th of August, 2004, at 12 noon. Helsinki University of Technology Department of Chemical Technology Laboratory of Bioprocess Engineering Teknillinen korkeakoulu Kemian osasto Bioprosessitekniikan laboratorio Distribution: Helsinki University of Technology Laboratory of Bioprocess Engineering P.O. Box 6100 FIN-02015 HUT Tel. +358-9-4512541 Fax. +358-9-462373 E-mail: [email protected] ©Antti Vuolanto ISBN 951-22-7176-1 (printed) ISBN 951-22-7177-X (pdf) ISSN 0359-6621 Espoo 2004 Vuolanto, Antti. Cross-linked protein crystal technology in bioseparation and biocatalytic applications. Espoo 2004, Helsinki University of Technology. Abstract Chemical cross-linking of protein crystals form an insoluble and active protein matrix. Cross-linked protein crystals (CLPCs) have many excellent properties including high volumetric activity and stability. In this thesis CLPC technology was studied in bioseparation and biocatalytic applications. A novel immunoaffinity separation material, cross-linked antibody crystals (CLAC), was developed in this thesis for enantiospecific separation of a chiral drug, finrozole. Previously, the preparation of an antibody Fab fragment ENA5His capable of enantiospecific affinity separation of the chiral drug has been described. -
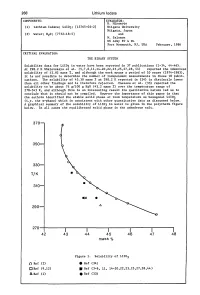
Evaluation of Aqueous Systems
268 Lithium Iodate COMPONENTS: EVALUATOR: H. Miyamoto (1) Lithium Iodate; LiI03; [13765-03-2] Niigata University Niigata, Japan (2) Water; H20; [7732-18-5] and M. Salomon US Army ET & DL Fort Monmouth, NJ, USA February, 1986 CRITICAL EVALUATION: THE BINARY SYSTEM Solubility data for LiI03 in water have been reported in 37 publications (1-34, 44-46). At 298.2 K Shklovskaya et al. (5,7,8,11,14-20,22,23,25,27,28,44) reported the identical solubility of 43.82 mass %, and although the work spans a period of 10 years (1974-1983), it is not possible to determine the number of independent measurements in these 18 publi cations. The solubility of 43.30 mass % at 298.2 K reported in (24) is distinctly lower than all other findings and is therefore rejected. Unezawa et al. (33) reported the solubility to be about 76 g/lOO g H20 (43.2 mass %) over the temperature range of 278-343 K, and although this is an interesting result its qualitative nature led us to conclude that it should not be compiled. However the importance of this paper is that the authors identified the stable solid phase at room temperature as hexagonal LiI03 (i.e. the a-phase) which is consistent with other quantitative data as discussed below. A graphical summary of the solubility of LiI03 in water is given in the polytherm figure below. In all cases the equilibrated solid phase is the anhydrous salt. 370 350 330 T/K 310 290 270-I------.r-----.-----..----......----------. 42 43 44 45 46 47 48 mass % Figure 1. -

University of Warwick Institutional Repository
University of Warwick institutional repository: http://go.warwick.ac.uk/wrap A Thesis Submitted for the Degree of PhD at the University of Warwick http://go.warwick.ac.uk/wrap/3994 This thesis is made available online and is protected by original copyright. Please scroll down to view the document itself. Please refer to the repository record for this item for information to help you to cite it. Our policy information is available from the repository home page. STUDIES OF HEXAHELICENE BONDED PHASES FOR THE HPLC RESOLUTION OF ENANTIOMERS by AURORA DEL ALAMO A thesis submitted in partial fulfilment of the requirements for the degreeof Doctor of Philosophy at the University of Warwick Department of Chemistry University of Warwick June, 1995 CONTENTS ChaDter One Pg LITERATURE REVIEW: CHIRAL LIQUID CHROMATOGRAPHY 1.1 Introduction 1 1.2 Resolution of Enantiomers 3 1.3 Preparation of FIPLC Chiral Stationary Phases 4 1.4 Development of Commercially Available 5 Direct Chiral Resolution Phases 1.4.1 Chiral Ligand Exchange Chromatography (LEC) 6 1.4.2 Synthetic Multiple Interaction CSPs 8 1.4.3 Protein Chiral Stationary Phases 13 1.4.4 Cyclodextrin CSPs 18 1.4.5 Polysaccharide Phases 31 1.4.6 Synthetic Polymer CSPs 36 1.4.7 Chiral Crown Ethers 40 Chapter Two LITERATURE REVIEW: SOME ASPECTS OF THE CHEMISTRY AND PROPERTIES OF HELICENES % 2.1 Helicenes 45 2.2 Synthesesof Hexahelicenes: Methods and their Problems 46 2.2.1 Non-Photochemical Syntheses 46 2.2.2 Photochemical Syntheses 48 2.2.3 Asymmetric Synthesesof Helicenes: 56 2.2.3.1 Asymmetric Synthesis -

Handbook on the Physics and Chemistry of Rare Earths Volume 9 Elsevier, 1987
Handbook on the Physics and Chemistry of Rare Earths volume 9 Elsevier, 1987 Edited by: Karl A. Gschneidner, Jr. and LeRoy Eyring ISBN: 978-0-444-87045-2 by kmno4 Handbook on the Physics and Chemistry of Rare Earths, edited by K.A. Gschneidner, Jr. and L. Eyring © Elsevier Science Publishers B.V., 1987 PREFACE Karl A. GSCHNEIDNER, Jr., and LeRoy EYRING o These elements perplex us in our rearches [sic], baffle us in our speculations, and haunt us in our very dreams. They stretch like an unknown sea before us- mocking, mystifying, and murmuring strange revelations and possibilities. Sir William Crookes (February 16, 1887) There are those who feel that the rare earth elements are destined to play an even greater role in our "high-tech" society in the future than they have in the past. This judgement is based upon the trend of increasing applications resulting from the electronic structures of these materials that lead to their unusual optical, magnetic, electrical and chemical properties so adaptable to the demands now being placed on materials. The "Handbook" seeks to provide topical reviews and compilations of critically reviewed data to aid in the design and fabrication of the required new materials. Four additional chapters in this tradition are the contents of this volume. The development of lasers containing rare earth species sparked an interest in glasses containing these elements. Reisfeld and J0rgensen discuss excited-state phenomena in vitreous rare-earth-containing substances. The description of the chemistry and crystal chemistry of complex inorganic compounds of the rare earths is continued by Niinist6 and Leskelfi from the previous volume. -

Stereoisomerism and Chirality Atin Shanker
IOSR Journal of Applied Chemistry (IOSR-JAC) e-ISSN: 2278-5736.Volume 14, Issue 6 Ser. I (June 2021), PP 10-12 www.iosrjournals.org Stereoisomerism and Chirality Atin Shanker Abstract The purpose of this study is to give a basic introduction to stereoisomerism and chirality. In this paper I discuss about the mechanisms involved behind their formation and their types. Chiral compounds play an important role in daily organic working. Homochirality, a controversial topic, is also discussed by showcasing its unconventional occurrence and results from a computer stimulated study by Chen Y, Ma W. Types of stereoisomers and how they occur in nature has also been discussed. This review will give you a basic idea about the opticality involved inorganic compounds specifically stereoisomers and its subsidiaries. --------------------------------------------------------------------------------------------------------------------------------------- Date of Submission: 14-06-2021 Date of Acceptance: 28-06-2021 --------------------------------------------------------------------------------------------------------------------------------------- I. Introduction Stereochemistry, Term originated c. 1878 by Viktor Meyer (1848–97) for the study of stereoisomers (see isomer). Louis Pasteur had shown in 1848 that tartaric acid has optical activity and that this depends on molecular asymmetry, and Jacobus H. van‟t Hoff and Joseph-Achille Le Bel (1847–1930) had independently explained in 1874 how a molecule with a carbon atom bonded to four different groups has two mirror-image forms. Stereochemistry deals with stereoisomers and with asymmetric synthesis. John Cornforth (b. 1917) and Vladimir Prelog (1906–98) shared a 1975 Nobel Prize for work on stereochemistry and stereoisomerism of alkaloids, enzymes, antibiotics, and other natural compounds. Beginning early in the 19th century, developments in crystallography, optics, and chemistry in France set the stage for the discovery of molecular chirality by Louis Pasteur in 1848. -

Enzyme Supported Crystallization of Chiral Amino Acids
ISBN 978-3-89336-715-3 40 Band /Volume Gesundheit /Health 40 Gesundheit Enzyme supported crystallization Health Kerstin Würges of chiral amino acids Mitglied der Helmholtz-Gemeinschaft Kerstin Würges Kerstin aminoacids Enzyme supported ofchiral crystallization Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 Forschungszentrum Jülich GmbH Institute of Bio- and Geosciences (IBG) Biotechnology (IBG-1) Enzyme supported crystallization of chiral amino acids Kerstin Würges Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 ISSN 1866-1785 ISBN 978-3-89336-715-3 Bibliographic information published by the Deutsche Nationalbibliothek. The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available in the Internet at http://dnb.d-nb.de. Publisher and Forschungszentrum Jülich GmbH Distributor: Zentralbibliothek 52425 Jülich Phone +49 (0) 24 61 61-53 68 · Fax +49 (0) 24 61 61-61 03 e-mail: [email protected] Internet: http://www.fz-juelich.de/zb Cover Design: Grafische Medien, Forschungszentrum Jülich GmbH Printer: Grafische Medien, Forschungszentrum Jülich GmbH Copyright: Forschungszentrum Jülich 2011 Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 D 61 (Diss. Düsseldorf, Univ., 2011) ISSN 1866-1785 ISBN 978-3-89336-715-3 The complete volume ist freely available on the Internet on the Jülicher Open Access Server (JUWEL) at http://www.fz-juelich.de/zb/juwel Neither this book nor any part of it may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage and retrieval system, without permission in writing from the publisher.