Structural and Functional Characterization of Neuraminidase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Connecting Bangladesh: Economic Corridor Network
Connecting Bangladesh: Economic Corridor Network Economic corridors are anchored on transport corridors, and international experience suggests that the higher the level of connectivity within and across countries, the higher the level of economic growth. In this paper, a new set of corridors is being proposed for Bangladesh—a nine-corridor comprehensive integrated multimodal economic corridor network resembling the London Tube map. This paper presents the initial results of the research undertaken as an early step of that development effort. It recommends an integrated approach to developing economic corridors in Bangladesh that would provide a strong economic foundation for the construction of world-class infrastructure that, in turn, could support the growth of local enterprises and attract foreign investment. About the Asian Development Bank COnnecTING BANGLADESH: ADB’s vision is an Asia and Pacific region free of poverty. Its mission is to help its developing member countries reduce poverty and improve the quality of life of their people. Despite the region’s many successes, it remains home to a large share of the world’s poor. ADB is committed to reducing poverty through inclusive economic growth, environmentally sustainable growth, and regional integration. ECONOMIC CORRIDOR Based in Manila, ADB is owned by 67 members, including 48 from the region. Its main instruments for helping its developing member countries are policy dialogue, loans, equity investments, guarantees, grants, NETWORK and technical assistance. Mohuiddin Alamgir -

Road Investment Strategy 2: 2020-2025
Road Investment Strategy 2: 2020–2025 March 2020 CORRECTION SLIP Title: Road Investment Strategy 2: 2020-25 Session: 2019-21 ISBN: 978-1-5286-1678-2 Date of laying: 11th March 2020 Correction: Removing duplicate text on the M62 Junctions 20-25 smart motorway Text currently reads: (Page 95) M62 Junctions 20-25 – upgrading the M62 to smart motorway between junction 20 (Rochdale) and junction 25 (Brighouse) across the Pennines. Together with other smart motorways in Lancashire and Yorkshire, this will provide a full smart motorway link between Manchester and Leeds, and between the M1 and the M6. This text should be removed, but the identical text on page 96 remains. Correction: Correcting a heading in the eastern region Heading currently reads: Under Construction Heading should read: Smart motorways subject to stocktake Date of correction: 11th March 2020 Road Investment Strategy 2: 2020 – 2025 Presented to Parliament pursuant to section 3 of the Infrastructure Act 2015 © Crown copyright 2020 This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/ open-government-licence/version/3. Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned. This publication is available at https://www.gov.uk/government/publications. Any enquiries regarding this publication should be sent to us at https://forms.dft.gov.uk/contact-dft-and-agencies/ ISBN 978-1-5286-1678-2 CCS0919077812 Printed on paper containing 75% recycled fibre content minimum. Printed in the UK by the APS Group on behalf of the Controller of Her Majesty’s Stationery Office. -

Module (Daughter Bd) 02-Nov-2015
8 7 6 5 4 3 2 1 THIS DRAWING IS THE PROPERTY OF ANALOG DEVICES INC. IT IS JUMPER TABLE REVISIONS NOT TO BE REPRODUCED OR COPIED, IN WHOLE OR IN PART, OR REV DESCRIPTION DATE APPROVED USED IN FURNISHING INFORMATION TO OTHERS, OR FOR ANY OTHER JP# ON OFF A ORIGINATION NHR-039086 JUN-2014 R.GETZ PURPOSE DETRIMENTAL TO THE INTERESTS OF ANALOG DEVICES. THE EQUIPMENT SHOWN HEREON MAY BE PROTECTED BY PATENTS 1 OWNED OR CONTROLLED BY ANALOG DEVICES. 2 3 RELAY CONTROL CHART D 4 D CONTROL CODE DEVICE FUNCTION CONNECTOR 5 * SEE ASSEMBLY INSTRUCTIONS C C B B 02-NOV-2015 MODULE (DAUGHTER BD) TEMPLATE ENGINEER DATE ANALOG - HARDWARE SERVICES SCHEMATIC DEV CES - HARDWARE SYSTEMS HW TYPE : MODULE - Product(s): Zynq / AD9361 TEST ENGINEER <PRODUCT_1> - A COMPONENT ENGINEER PACKAGE : PinCount-lead N/A Package-family A - : Pitch-pitch StyleVendor Style TEST PROCESS <User Define> - HARDWARE RELEASE <User Define> - <User Define> DESIGNER MASTER PROJECT TEMPLATE TESTER TEMPLATE DRAWING NO. REV. S.LEE/ TBD no_template PTD ENGINEER UNLESS OTHERWISE SPECIFIED DIMENSIONS ARE IN INCHES TOLERANCES 02_038702 C <PTD_ENGINEER> DECIMALS FRACTIONS ANGLES P.O SPEC. BK/BD SPEC. SOCKET OEM OEM PART# HANDLER CHECKER SIZE SCALE CODE ID NO. X.XX +-0.010 +-1/32 +-2 - X.XXX +-0.005 DD 1:1 CodeID SHEET 1 OF 10 8 7 6 5 4 3 2 1 8 7 6 5 4 3 2 1 REVISIONS REV DESCRIPTION DATE APPROVED PHY1_1V8 3.3V E16 PHY1_VDD_3V3 PS_MIO17_501_ETH0_TX_D0 PS_MIO26_501_ETH0_RX_D3 PHY1_AVDD_1V8_OUT PS_MIO18_501_ETH0_TX_D1 PS_MIO25_501_ETH0_RX_D2 1 2 PHY1_AVDD_1V8 E13 L0805 1 2 PS_MIO19_501_ETH0_TX_D2 -
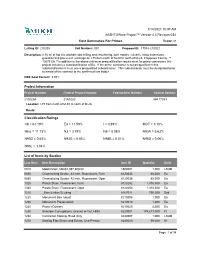
Aashtoware Project™ Version 4.4 Revision 034 1/14/2021 10:37 AM
1/14/2021 10:37 AM AASHTOWare Project™ Version 4.4 Revision 034 Cost Summaries For Primes Report v1 Letting ID: 210205 Call Number: 001 Proposal ID: 17033-210022 Description: 8.30 mi of hot mix asphalt cold milling and resurfacing, joint repairs, culverts, ramp extensions, guardrail and pavement markings on I-75 from north of M-80 to north of M-28, Chippewa County. ** 12075 Cb **In addition to the above minimum prequalification requirement for prime contractors this project includes a subclassification of Ea. If the prime contractor is not prequalified in this subclassification it must use a prequalified subcontractor. This subcontractor must be designated prior to award of the contract to the confirmed low bidder. DBE Goal Percent: 3.00% Project Information Project Number Federal Project Number Federal Item Number Control Section 210022A 21A0220 NH 17033 Location: I-75 from north of M-80 to north of M-28. Route: Classification Ratings Cb = 61.19% Ea = 11.59% I = 0.89% MOT = 3.10% Misc = 11.73% N3 = 3.79% N6 = 0.08% N93A = 5.63% N93D = 0.05% N93G = 0.85% N94B = 0.01% N95D = 0.06% N96L = 1.04% List of Items by Section Line Num Item Description Item ID Quantity Units 0010 Mobilization, Max$1,097,800.00 1500001 1.000 LSUM 0850 Channelizing Device, 42 inch, Fluorescent, Furn 8120035 65.000 Ea 0860 Channelizing Device, 42 inch, Fluorescent, Oper 8120036 65.000 Ea 1020 Plastic Drum, Fluorescent, Furn 8120252 1,310.000 Ea 1030 Plastic Drum, Fluorescent, Oper 8120253 1,310.000 Ea 1210 _Bioretention Seeding 8167011 750.000 Syd 1220 Monument Box -

Powering an I.MX 6SX-Based System Using the PF3000 PMIC
NXP Semiconductors Document Number: AN5161 Application Note Rev. 1.0, 7/2016 Powering an i.MX 6SX-based system using the PF3000 PMIC 1 Introduction Contents 1 Introduction. 1 The PF3000 is a highly integrated Power Management IC ideally suited to power NXP's i.MX 6SX, i.MX 6SL, i.MX 6S, i.MX 6DL, 2 PF3000 voltage regulators . 2 i.MX 6UL and the i.MX 7 series of applications processors. This 3 i.MX 6SX power management using the PF3000 . 3 Application Note discusses the power tree configuration for 4 PF3000 power input . 4 powering an i.MX 6SX based system using the PF3000. NXP analog ICs are manufactured using the SMARTMOS 5 PF3000 layout guidelines . 5 process, a combinational BiCMOS manufacturing flow that 6 PF3000 software support . 5 integrates precision analog, power functions and dense CMOS 7 Schematics. 6 logic together on a single cost-effective die. 8 References . 27 9 Revision history . 28 © 2016 NXP B.V. PF3000 voltage regulators 2 PF3000 voltage regulators Table 1 shows a summary of the voltage regulators in the PF3000. Output voltage and startup sequence of the regulators is programmed into the PMIC through One Time Programmable (OTP) memory. For more details, refer to the product datasheet. Table 1. PF3000 voltage regulators Regulator Output voltage range Load current rating 0.7 V to 1.425 V SW1A 1.8 V 1000 mA 3.3 V SW1B 0.7 V to 1.475 V 1750 mA 1.5 V to 1.85 V SW2 1250 mA 2.5 V to 3.3 V SW3 0.9 V to 1.65 V 1500 mA SWBST 5.0 V to 5.15 V 600 mA VSNVS 3.0 V 1.0 mA VLDO1 1.8 V to 3.3 V 100 mA VLDO2 0.8 V to 1.55 V 250 mA VLDO3 1.8 V to 3.3 V 100 mA VLDO4 1.8 V to 3.3 V 350 mA 1.8 V to 1.85 V VCC_SD 100 mA 2.85 V to 3.3 V V33 2.85 V to 3.3 V 350 mA VREFDDR VINREFDDR/2 10 mA Powering an i.MX 6SX-based system using the PF3000 PMIC, Rev. -

Grand Trunk Road Improvement Project
NATIONALHIGHWAYS AUTHORITY OFINDIA NotMm Trmk ROL" in Mncit inba (bp( M&%p&uryon) Public Disclosure Authorized Grand Trunk Road Improvement Project Environmentl Management Plan Package IVA t \' ~~N4rChzT">k Rouh5G E432 C -, Volume 10 Public Disclosure Authorized Northws Tnk Read r- 1I O iCeftury 'a , ,¢,,-t4 (G.T. Road) ;-Robad 1`44twofit S t s. / Public Disclosure Authorized l ; /h-r r' d (2007aA.d.) Propoead TrUOl4NU Mpoi -~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~- Public Disclosure Authorized / ~~FIL1E GOPY i ; _~ ss Siouth Asia Pvt. 'Ltd.- -- _ AQVS WJ 99 ........-----------..-.... .Nd SaN.VaNv 1nuinoLLsoH 5 S.C LI 99.................................... I................................................................... NION3 1 4 VICv 1, S9... --- --- ------- ------ '-'-'S3UUiOnW.S NOIUV9 ZVCL' pg.....................-........ I.......-..............----.--.-----.....-''........ ONIUM d0N'33 1 19 .-.--.--..-- ---- --- ----- --- - - W3O803NI3j B'£V' I 85'9 ',.. ,..... .------- '-'''-'-(DNWILLlH9 VHV NIELN~3o)WOUM flS0ISC 8 £ I 99. .............................-.....................-----..-..............................nS NOM Y3ON03 L£ i, -- 'oSM .S---- SMIDISI 9~£ I .ZSilO ~~~~3soo~~~~~00dI~~~~~ dYwvc1 SC £ I 19 ... s~3juINmB~ Z£C 1 Z9., . I........"I,,'-",.. - - - ........- b1V'-'sMS-flre VCV i, .- _ _ - OM-J.N3VE14IN WJN3V4NOAEN3 U0 SNLVOIO CII 94..n-------------------------- SIN3V31N33N*f~VN3 1VldAj -- Z. [K[ s., ... .. ...... .. ... .-......- ......... sno100W dN Zs -................................................................- ONMON -

Biological Properties and Genetic Characterization of Novel Low
microorganisms Communication Biological Properties and Genetic Characterization of Novel Low Pathogenic H7N3 Avian Influenza Viruses Isolated from Mallard Ducks in the Caspian Region, Dagestan, Russia Marina Gulyaeva 1,2,* , Maria Alessandra De Marco 3, Ganna Kovalenko 4, Eric Bortz 4 , Tatiana Murashkina 1, Kseniya Yurchenko 1 , Marzia Facchini 5, Mauro Delogu 6, Ivan Sobolev 1, Alimurad Gadzhiev 7, Kirill Sharshov 1 and Alexander Shestopalov 1 1 Federal State Budget Scientific Institution, Federal Research Center of Fundamental and Translational Medicine, 630117 Novosibirsk, Russia; [email protected] (T.M.); [email protected] (K.Y.); [email protected] (I.S.); [email protected] (K.S.); [email protected] (A.S.) 2 Department of Natural Science, Novosibirsk State University, 630090 Novosibirsk, Russia 3 ISPRA Institute for Environmental Protection and Research, 40064 Ozzano dell’Emilia (BO), Italy; [email protected] 4 Department of Biological Sciences, University of Alaska, Anchorage, AK 99508, USA; [email protected] (G.K.); [email protected] (E.B.) 5 Department of Infectious Diseases, Istituto Superiore di Sanità, 00161 Rome, Italy; [email protected] 6 Department of Veterinary Medical Sciences, University of Bologna, 40064 Ozzano dell’Emilia (BO), Italy; Citation: Gulyaeva, M.; De Marco, [email protected] M.A.; Kovalenko, G.; Bortz, E.; 7 Department of Ecology and Sustainable Development, Dagestan State University, 367000 Dagestan, Russia; Murashkina, T.; Yurchenko, K.; [email protected] Facchini, M.; Delogu, M.; Sobolev, I.; * Correspondence: [email protected]; Tel.: +7-(952)-913-65-13 Gadzhiev, A.; et al. Biological Properties and Genetic Abstract: Avian influenza viruses (AIVs) are maintained in wild bird reservoirs, particularly in Characterization of Novel Low mallard ducks and other waterfowl. -
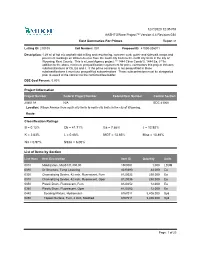
Classification Ratings Project Information B = 0.12% Cb = 41.71
12/7/2020 12:35 PM AASHTOWare Project™ Version 4.4 Revision 034 Cost Summaries For Primes Report v1 Letting ID: 210108 Call Number: 001 Proposal ID: 41000-206011 Description: 1.29 mi of hot mix asphalt cold milling and resurfacing, concrete curb, gutter and sidewalk ramps and pavement markings on Wilson Avenue from the south city limits to the north city limits in the city of Wyoming, Kent County. This is a Local Agency project. ** 1444 Cb or Comb/Jt. 1444 Ea, J **In addition to the above minimum prequalification requirement for prime contractors this project includes subclassifications of Cb, Ea and J. If the prime contractor is not prequalified in those subclassifications it must use prequalified subcontractors. Those subcontractors must be designated prior to award of the contract to the confirmed low bidder. DBE Goal Percent: 0.00% Project Information Project Number Federal Project Number Federal Item Number Control Section 206011A N/A EDC 41000 Location: Wilson Avenue from south city limits to north city limits in the city of Wyoming. Route: Classification Ratings B = 0.12% Cb = 41.71% Ea = 7.86% J = 12.92% K = 2.83% L = 0.48% MOT = 13.65% Misc = 13.46% N3 = 0.97% N93A = 6.00% List of Items by Section Line Num Item Description Item ID Quantity Units 0010 Mobilization, Max$131,300.00 1500001 1.000 LSUM 0090 Dr Structure, Temp Lowering 4030390 44.000 Ea 0300 Channelizing Device, 42 inch, Fluorescent, Furn 8120035 250.000 Ea 0310 Channelizing Device, 42 inch, Fluorescent, Oper 8120036 250.000 Ea 0350 Plastic Drum, Fluorescent, Furn -
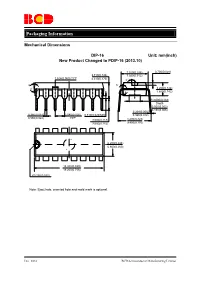
Mechanical-Dimensions-BCD.Pdf
Packaging Information Mechanical Dimensions DIP-16 Unit: mm(inch) New Product Changed to PDIP-16 (2013.10) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060) TYP 4.310(0.170) 5° 6° 6° 4° 3.200(0.126) 4° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014) 0.560(0.022) TYP 3.000(0.118) 8.200(0.323) 3.600(0.142) 9.400(0.370) 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optional. Dec. 2014 BCD Semiconductor Manufacturing Limited Packaging Information Mechanical Dimensions DIP-16 (Special for AZ4052) Unit: mm(inch) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060)T YP 4.310(0.170) 5 ° 6 ° 6 ° 4 ° 3.200(0.126) 4 ° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014 ) 0.560(0.022) TYP 3.000(0.118) 8.500(0.335) 3.600(0.142) TYP 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optiona.l Dec. -

Notebook Computer M350C/M360C Service Manual Preface
Preface Notebook Computer M350C/M360C Service Manual Preface I Preface Notice The company reserves the right to revise this publication or to change its contents without notice. Information contained herein is for reference only and does not constitute a commitment on the part of the manufacturer or any subsequent ven- dor. They assume no responsibility or liability for any errors or inaccuracies that may appear in this publication nor are they in anyway responsible for any loss or damage resulting from the use (or misuse) of this publication. This publication and any accompanying software may not, in whole or in part, be reproduced, translated, transmitted or reduced to any machine readable form without prior consent from the vendor, manufacturer or creators of this publica- tion, except for copies kept by the user for backup purposes. Brand and product names mentioned in this publication may or may not be copyrights and/or registered trademarks of their respective companies. They are mentioned for identification purposes only and are not intended as an endorsement of that product or its manufacturer. Version 1.0 July 2003 Preface Trademarks Intel® and Pentium® are registered trademarks of Intel Corporation. Windows® is a registered trademark of Microsoft Corporation. Other brand and product names are trademarks and./or registered trademarks of their respective companies. II Preface About this Manual This manual is intended for service personnel who have completed sufficient training to undertake the maintenance and inspection of personal computers. It is organized to allow you to look up basic information for servicing and/or upgrading components of the M350C/ M360C series notebook PC. -

Etruscan Glossarya.Pdf
A B CD 1 2 Note: This glossary supplements Table 1. Copyright © 1981-2017 Mel Copeland. All rights reserved. 3 4.27.17 - Items in red are changes; often updated For those using the PDF version of this file, see the latest changes Etruscan_GlossaryA.xls at Etruscan Phrases 4 "X" locators designate Anatolian (Phrygian) texts 5 Updated to reconcile declension patterns 6 http://www.maravot.com/Etruscan_Phrases_a.html 7 Contact: [email protected] 8 9 English Etruscan Location 10 to, in (L. a) A TC120,TC127, Au95, Au102, AG-2, Z92, AN12, AN100, 11 to, in (L. a) A (continued) N21, N206, N371,Q701, Q717, R381, R499, N722, N731, MS23 12 to, in (L. a) A (continued) Q376, Q388, R542, R584, AH-9, AC-3, TC211, K159, PJ-1, J5-6, J40-10, PV-7, PW-12 13 and, and also, and indeed (L. ac, atque) AC Z58, Z432, Z1183, Au-1, TC46, TC90, Au95, K149, L50, J41-2 14 and, and also, and indeed (L. ac, atque) AK Z489, Z508, Z1139, XQ-1 15 call, to (L. accio-aire) ACA Z572, TC46 16 it/he will move, set in motion (L. ago-agere, Ind. I Fut. 3rd Pers. Single aget) ACE J40-8 17 call, to (L. accio-aire) ACeR M71 18 prophesy, to wish (L. auguro-are) ACERN (they prophesy) DL-2 (This mirror depicts reading from a liver) 19 level, make equal, compare (L. acquo-are) ACES N462 20 Achaia? (L. Achaia or Achaia-ae, Achaia or in Gen. Greece) ACHIE (AKIE) CP35 21 Agememnon ACHMEMNVN DM-6, CG-3 22 Achilles – see CG-1 ACHLE (AKLE) (See ACHVLE) MM-5, CG-1, DP-1, LM-4? CCG-3 23 Achilles – see CG-1 ACHL or ACHLA (ACH LA) CH-2 24 Achule, god in company of Thetis on a mirror, probably Achilles ACHVLE (AKVLE) (See ACHLE) CQ-2 25 Achloser, name of Briseis, concubine of Achilles? ACHLVSR ( ACHLPIMSR?, ACHVPIMSR?) CQ3 26 call, to (L. -
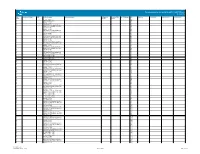
Pin Information for the Intel® Arria® 10 10AX115 Device Version 1.6
Pin Information for the Intel® Arria® 10 10AX115 Device Version 1.6 Bank Index within I/O Bank (1) VREF Pin Name/Function Optional Function(s) Configuration Dedicated Tx/Rx Soft CDR Support HF34 DQS for X4 DQS for X8/X9 DQS for X16/X18 DQS for X32/X36 Number Function Channel 1F REFCLK_GXBL1F_CHTp M28 1F REFCLK_GXBL1F_CHTn M27 1F GXBL1F_TX_CH5n B31 1F GXBL1F_TX_CH5p B32 1F GXBL1F_RX_CH5n,GXBL1F_REFCLK5n C29 1F GXBL1F_RX_CH5p,GXBL1F_REFCLK5p C30 1F GXBL1F_TX_CH4n D31 1F GXBL1F_TX_CH4p D32 1F GXBL1F_RX_CH4n,GXBL1F_REFCLK4n E29 1F GXBL1F_RX_CH4p,GXBL1F_REFCLK4p E30 1F GXBL1F_TX_CH3n F31 1F GXBL1F_TX_CH3p F32 1F GXBL1F_RX_CH3n,GXBL1F_REFCLK3n G29 1F GXBL1F_RX_CH3p,GXBL1F_REFCLK3p G30 1F GXBL1F_TX_CH2n H31 1F GXBL1F_TX_CH2p H32 1F GXBL1F_RX_CH2n,GXBL1F_REFCLK2n J29 1F GXBL1F_RX_CH2p,GXBL1F_REFCLK2p J30 1F GXBL1F_TX_CH1n C33 1F GXBL1F_TX_CH1p C34 1F GXBL1F_RX_CH1n,GXBL1F_REFCLK1n K31 1F GXBL1F_RX_CH1p,GXBL1F_REFCLK1p K32 1F GXBL1F_TX_CH0n E33 1F GXBL1F_TX_CH0p E34 1F GXBL1F_RX_CH0n,GXBL1F_REFCLK0n L29 1F GXBL1F_RX_CH0p,GXBL1F_REFCLK0p L30 1F REFCLK_GXBL1F_CHBp P28 1F REFCLK_GXBL1F_CHBn P27 1E REFCLK_GXBL1E_CHTp T28 1E REFCLK_GXBL1E_CHTn T27 1E GXBL1E_TX_CH5n G33 1E GXBL1E_TX_CH5p G34 1E GXBL1E_RX_CH5n,GXBL1E_REFCLK5n M31 1E GXBL1E_RX_CH5p,GXBL1E_REFCLK5p M32 1E GXBL1E_TX_CH4n J33 1E GXBL1E_TX_CH4p J34 1E GXBL1E_RX_CH4n,GXBL1E_REFCLK4n N29 1E GXBL1E_RX_CH4p,GXBL1E_REFCLK4p N30 1E GXBL1E_TX_CH3n L33 1E GXBL1E_TX_CH3p L34 1E GXBL1E_RX_CH3n,GXBL1E_REFCLK3n P31 1E GXBL1E_RX_CH3p,GXBL1E_REFCLK3p P32 1E GXBL1E_TX_CH2n N33 1E GXBL1E_TX_CH2p N34 1E GXBL1E_RX_CH2n,GXBL1E_REFCLK2n