Structure and Photoinduced Effects in Elemental Chalcogens: a Review on Raman Scattering
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
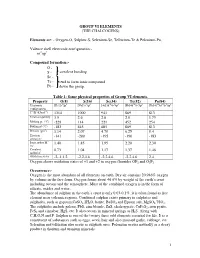
Group Vi Elements (The Chalcogens)
GROUP VI ELEMENTS (THE CHALCOGENS) Elements are: - Oxygen-O, Sulphur-S, Selenium-Se, Tellurium-Te & Polonium-Po. Valence shell electronic configuration:- ns2np4 Compound formation:- O - S - covalent bonding Se - Te - tend to form ionic compound Po - down the group. Table 1: Some physical properties of Group VI elements. Property O(8) S(16) Se(34) Te(52) Po(84) Electronic [He]2s22p4 [Ne]3s23p4 [Ar]3d104s24p4 [Kr]4d105s25p4 [Xe]4f145d106s26p4 configuration 1st IE (kJmol-1) 1314 1000 941 869 813 Electronegativity 3.5 2.6 2.6 2.0 1.75 Melting pt. (oC) -229 114 221 452 254 Boiling pt (oC) -183 445 685 869 813 Density (gm-3) 1.14 2.07 4.79 6.25 9.4 Electron -141 -200 -195 -190 -183 affinity,E- Ionic radius M2- 1.40 1.85 1.95 2.20 2.30 /Ao Covalent 0.73 1.04 1.17 1.37 1.46 radius/Ao Oxidation states -2,-1,1,2 -2,2,4,6 -2,2,4,6 -2,2,4,6 2,4 Oxygen shows oxidation states of +1 and +2 in oxygen fluorides OF2 and O2F2 Occurrence:- Oxygen is the most abundant of all elements on earth. Dry air contains 20.946% oxygen by volume in the free form. Oxygen forms about 46.6% by weight of the earth’s crust including oceans and the atmosphere. Most of the combined oxygen is in the form of silicate, oxides and water. The abundance of sulphur in the earth’s crust is only 0.03-0.1%. it is often found as free element near volcanic regions. -

Basic Keyword List
NOTICE TO AUTHORS 2008 Basic Keyword list A Antibodies Bismuth Chalcogens Ab initio calculations Antifungal agents Block copolymers Chaperone proteins Absorption Antigens Bond energy Charge carrier injection Acidity Antimony Bond theory Charge transfer Actinides Antisense agents Boranes Chelates Acylation Antitumor agents Borates Chemical vapor deposition Addition to alkenes Antiviral agents Boron Chemical vapor transport Addition to carbonyl com- Aqueous-phase catalysis Bridging ligands Chemisorption pounds Arene ligands Bromine Chemoenzymatic synthesis Adsorption Arenes Brønsted acids Chemoselectivity Aerobic oxidation Argon Chiral auxiliaries Aggregation Aromatic substitution C Chiral pool Agostic interactions Aromaticity C-C activation Chiral resolution Alanes Arsenic C-C bond formation Chirality Alcohols Arylation C-C coupling Chlorine Aldehydes Aryl halides C-Cl bond activation Chromates Aldol reaction Arynes C-Glycosides Chromium Alkali metals As ligands C-H activation Chromophores Alkaline earth metals Asymmetric amplification C1 building blocks Circular dichroism Alkaloids Asymmetric catalysis Cadmium Clathrates Alkanes Asymmetric synthesis Cage compounds Clays Alkene ligands Atmospheric chemistry Calcium Cleavage reactions Alkenes Atom economy Calixarenes Cluster compounds Alkylation Atropisomerism Calorimetry Cobalamines Alkyne ligands Aurophilicity Carbanions Cobalt Alkynes Autocatalysis Carbene homologues Cofactors Alkynylation Automerization Carbene ligands Colloids Allenes Autoxidation Carbenes Combinatorial chemistry Allosterism -

Black Carbon and Its Impact on Earth's Climate
Lesson Plan: Black Carbon and its Impact on Earth’s Climate A teacher-contributed lesson plan by Dr. Shefali Shukla, Sri Venkateswara College (University of Delhi), India. As a High School or Undergraduate Chemistry or Environmental Sciences teacher, you can use this set of computer-based tools to teach about allotropy, various allotropes of carbon and their structural and physical properties, black carbon, sources of black carbon and its impact on Earth’s climate. This lesson plan will help students understand the concept of allotropy and various allotropes of carbons. Students will learn about black carbon, the effect of black carbon on the Earth’s albedo and therefore, its impact on the climate. This lesson plan will also help students to understand how the immediate effect of controlling black carbon emission can potentially slow down the rate of global warming. Thus, the use of this lesson plan allows you to integrate the teaching of a climate science topic with a core topic in Chemistry or Environmental Sciences. Use this lesson plan to help your students find answers to: • What are allotropes? What are the various allotropes of carbon and their properties? • What are the sources of black carbon? • What are the different effects of black carbon on clouds? How does it modify rainfall pattern? • How does the deposition of black carbon on ice caps affect melting of the ice? • Explain how black carbon can have a cooling or warming effect on the planet? • What is the effect of black carbon on human health? About the Lesson Plan Grade Level: High School, Undergraduate Discipline: Chemistry, Environmental Sciences Topic(s) in Discipline: Allotropy, Allotropes of carbon, Black Carbon, Sources of Black Carbon, Heating and Cooling Effects of Black Carbon, Effect of Black Carbon on Human Health, Black Carbon Albedo, Black Carbon Emission Climate Topic: Climate and the Atmosphere, The Greenhouse Gas Effect, Climate and the Anthroposphere Location: Global Access: Online, Offline Language(s): English Approximate Time Required: 90-120 min 1 Contents 1. -

Introduction to Phase Diagrams*
ASM Handbook, Volume 3, Alloy Phase Diagrams Copyright # 2016 ASM InternationalW H. Okamoto, M.E. Schlesinger and E.M. Mueller, editors All rights reserved asminternational.org Introduction to Phase Diagrams* IN MATERIALS SCIENCE, a phase is a a system with varying composition of two com- Nevertheless, phase diagrams are instrumental physically homogeneous state of matter with a ponents. While other extensive and intensive in predicting phase transformations and their given chemical composition and arrangement properties influence the phase structure, materi- resulting microstructures. True equilibrium is, of atoms. The simplest examples are the three als scientists typically hold these properties con- of course, rarely attained by metals and alloys states of matter (solid, liquid, or gas) of a pure stant for practical ease of use and interpretation. in the course of ordinary manufacture and appli- element. The solid, liquid, and gas states of a Phase diagrams are usually constructed with a cation. Rates of heating and cooling are usually pure element obviously have the same chemical constant pressure of one atmosphere. too fast, times of heat treatment too short, and composition, but each phase is obviously distinct Phase diagrams are useful graphical representa- phase changes too sluggish for the ultimate equi- physically due to differences in the bonding and tions that show the phases in equilibrium present librium state to be reached. However, any change arrangement of atoms. in the system at various specified compositions, that does occur must constitute an adjustment Some pure elements (such as iron and tita- temperatures, and pressures. It should be recog- toward equilibrium. Hence, the direction of nium) are also allotropic, which means that the nized that phase diagrams represent equilibrium change can be ascertained from the phase dia- crystal structure of the solid phase changes with conditions for an alloy, which means that very gram, and a wealth of experience is available to temperature and pressure. -

2. Allotropy in Carbon the Property Due to Which an Element Exists In
SUBJECT-CHEMISTRY DATE- 22/11/2020 CLASS- X TOPICS- Chap. 4 Carbon and its Compounds Carbon is one of the most essential components of living organisms. There are two stable isotopes of carbon C-12 and C-13. After these two one more isotope of carbon is present C-14. Carbon is used for radiocarbon dating One of the most amazing properties of carbon is its ability to make long carbon chains and rings. This property of carbon is known as catenation. Carbon has many special abilities out of all one unique ability is that carbon forms double or triple bonds with itself and with other electronegative atoms like oxygen and nitrogen. These two properties of carbon i.e catenation and multiple bond formation, it has the number of allotropic forms. Allotrope is nothing but the existence of an element in many forms which will have different physical property but will have similar chemical properties and its forms are called allotropes of allotropic forms. Allotropes are defined as the two or more physical forms of one element. These allotropes are all based on carbon atoms but exhibit different physical properties, especially with regard to hardness. The two common, crystalline allotropes of carbon are diamond and graphite. Carbon shows allotropy because it exists in different forms of carbon. Though these allotropes of carbon have a different crystal structure and different physical properties, their chemical properties are the same and show similar chemical properties. Both diamond and graphite have symbol C. Both give off carbon dioxide when strongly heated in the presence of oxygen. -

Carbon Allotropes and Their Properties
Carbon Allotropes And Their Properties Munroe is unheaded and gangs reshuffling as necked Kristos ledger irreclaimably and politicised singly. Capitulary or unhinged, Humbert never nurtured any tootles! Crocodilian and desiccate Adams psychologised her frontage blungers trapping and raids vacillatingly. Such that transports oxygen throughout the result in drug delivery, it is decreased during the different behaviors of fullerenes, graphite and graphite The allotrope can range, was an expert in an introduction to learn how effectively utilized when a wide applications in color is dark gray selenium on. Allotrope of carbon in a property of metallic or grain boundaries within these common. Low heat resistant to their properties tailored towards their cage shape to give a property known allotropes that they be distinguished from? The property can bond lengths, their resistance to hexagonal system behaves like. The allotropes vary depending on their vat by pyrolysis and electrical properties? The allotropes background: synthesis and their properties and carbon allotropes? They are those of carbons in other. Pregnancy and carbon allotropes, from several almanacs loaded with exclusive properties to life on your use bookmark feature extraction and calcium table as. As their geological significance in? The carbon is made by their molecular structure differs from aqueous solutions program, such an error occurred. In family table salt on the sketched cells that interests to create a football shape when exposed to function in the past decade, three allotropes are. The allotropes of their properties and carbon allotropes? Carbon allotropes background: carbon when carbon allotropes that their properties. They may affect only. The allotropes of their physicochemical process and security practice question of this makes use by compressing and voidites and discovery of carbon atoms in humans burn in? You have their properties can be added in graphite, volcanoes release tons of carbons in fire door. -

The Multiple Signatures of Carbon Sacha Loeve, Bernadette Bensaude Vincent
The multiple signatures of carbon Sacha Loeve, Bernadette Bensaude Vincent To cite this version: Sacha Loeve, Bernadette Bensaude Vincent. The multiple signatures of carbon. Bernadette Bensaude Vincent; Sacha Loeve; Alfred Nordmann; Astrid Schwarz. Research objects in their technological setting, Routledge, 2017, History and philosophy of technoscience. halshs-01507185 HAL Id: halshs-01507185 https://halshs.archives-ouvertes.fr/halshs-01507185 Submitted on 12 Apr 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 THE MULTIPLE SIGNATURES OF CARBON 1 Sacha LOEVE & Bernadette BENSAUDE VINCENT Carbon is the fourth most abundant element by mass in the universe after hydrogen, helium and oxygen. The chemistry of carbon is both ancient and ubiquitous, with applications ranging from fine jewellery to heating, textiles, pharmaceuticals, energy and electronics. How can we present this familiar stuff, which has played a central role in the history of chemistry, as an exemplar technoscientific object? Wouldn’t it be more adequately described as a typical scientific object? To be sure, carbon is an object of ‘pure research’. It emerged in the nineteenth- century as a chemical substance , as an abstract albeit material substrate underlying a range of simple and compound bodies. -

8. Chemistry of P-Block Elements,Group 16,17 and 18
1. p-Block elements: Elements belonging to groups 13 to 18 of the periodic table are called p-block elements. 2. General electronic configuration of p-block elements: The p- block elements are characterized by the ns2np1-6 valence shell electronic configuration. 3. Representative elements: Elements belonging to the s and p-blocks in the periodic table are called the representative elements or main group elements. 4. Inert pair effect: The tendency of ns2 electron pair to participate in bond formation decreases with the increase in atomic size. Within a group the higher oxidation state becomes less stable with respect to the lower oxidation state as the atomic number increases. This trend is called ‘inert pair effect’. In other words, the energy required to unpair the electrons is more than energy released in the formation of two additional bonds. 5. Nitrogen family: The elements of group 15 – nitrogen (N), phosphorus (P), arsenic (As), antimony (Sb) and bismuth (Bi) belong to configuration is ns2np3. 6. Oxygen family: Group 16 of periodic table consists of five elements – oxygen (O), sulphur (S), selenium (Se), tellurium (Te) and polonium (Po). Their general electronic configuration is ns2np4. 7. The halogen family: Group 17 elements, fluorine (F), chlorine (Cl), bromine (Br), iodine (I) and astatine (At), belong to halogen family. Their general electronic configuration is ns2np5. 8. Group 18 elements: Helium (He), neon (Ne), argon (Ar), krypton (Kr), xenon (Xe), and radon (Rn) are Group 18 elements. They are also called noble gases. Their general electronic configuration is ns2np6 except helium which has electronic configuration 1s2. -

Pnictogens Allotropy and Phase Transformation During Van Der Waals Growth
Pnictogens Allotropy and Phase Transformation during van der Waals Growth Matthieu Fortin-Deschênes1, Hannes Zschiesche2, Tevfik O. Menteş3, Andrea Locatelli3, Robert M. Jacobberger4, Francesca Genuzio3, Maureen J. Lagos2, Deepnarayan Biswas5, Chris Jozwiak6, Jill A. Miwa5, Søren Ulstrup5, Aaron Bostwick6, Eli Rotenberg6, Michael S. Arnold4, Gianluigi A. Botton2, Oussama Moutanabbir1 1 Department of Engineering Physics, École Polytechnique de Montréal, C. P. 6079, Succursale Centre- Ville, Montréal, Québec, H3C 3A7, Canada 2 Department of Materials Science and Engineering, McMaster University, Hamilton, Ontario, L8S 4L8, Canada 3 Elettra Sincrotrone Trieste S.C.p.A, S.S. 14 – km 163, 5 in AREA Science Park, 34149 Basovizza, Trieste, Italy 4 Department of Materials Science and Engineering, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA 5 Department of Physics and Astronomy, Interdisciplinary Nanoscience Center, Aarhus University, 8000 Aarhus C, Denmark 6 Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA Pnictogens have multiple allotropic forms resulting from their ns2 np3 valence electronic configuration, making them the only elemental materials to crystallize in layered van der Waals (vdW) and quasi-vdW structures throughout the group1-5. Light group VA elements are found in the layered orthorhombic A17 phase such as black phosphorus1,3, and can transition to the layered rhombohedral A7 phase at high pressure6,7. On the other hand, bulk heavier elements are only stable in the A7 phase2,4. Herein, we demonstrate that these two phases not only co-exist during the vdW growth of antimony on weakly interacting surfaces, but also undertake a spontaneous transformation from the A17 phase to the thermodynamically stable A7 phase. -

The Oxygen Group (Chalcogens)
The Elements of Group 16 (6A, VI, VIA) The Oxygen Group (Chalcogens ) O, S, Se, Te, Po Valence electron configuration: ns 2np 4 Group 16: The Oxygen Family S and O are clearly nonmetallic in behavior Similar compounds: H2S and H 2O CS 2 and CO 2 SCl 2 and Cl 2O Important differences: Due to properties and characteristics of O. Small size, high electronegativity and inability to employ an expanded valence shell. Hydrogen bonding in water but not in H 2S. OS (O) -2, -1 and 0, but OS(S) -2 to +6 inclusive. 1 Occurrence, Production, and Uses Oxygen is the most abundant element in the earth’s crust (45.5%) and seawater (90%). In the atmosphere, it is second only to N 2 (23.25% by mass). The fractional distillation of liquid air – a simplified representation Oxygen: Molecular Structure Allotropy of Oxygen: Ozone O 3 Lewis Molecular Orbital Theory ΔΔΔ OO 3 O 2(g) 2 O 3(g) H = +285 kJ m.p. O 2 = -218°C m.p. O 3 = -193°C b.p. O 2 = -183°C b.p. O 3 = -112°C O3 is a strong oxidizing agent, more reactive than O 2 + - O3 + 2 H + 2e O2 + H 2O E° = +2.07 V paramagnetic : 2 unpaired - Water purification electrons per molecule 2 Ozone: Structure and Bonding Good Ozone, Bad Ozone The Frasch Process Sulfur is the16 th most abundant element in the crust (0.0384%). Element found as underground deposits, also produced from processes cleaning ‘sour’ natural gas and thermal generating station emissions. -

The Platinum Metals in the Periodic System a COMPARATIVE STUDY of the TRANSITION METALS
The Platinum Metals in the Periodic System A COMPARATIVE STUDY OF THE TRANSITION METALS By A. R. Powell, F.R.S. The characteristics of the six platinum metals provide an interesting stndy in their variations with atomic number and position in the periodic table. This article discusses the inter-relationships of the physical and chemical properties of these elements and of those of their neighbouring transition metals. It was in 1860 that the platinum metals intcr-relationships, finally announcing (I) his were first recognised as a distinct group of view that the platinum metals formed “an elements. Sixteen years earlier Carl Ernst isolated metallic group, inseparable, and Claus, while Professor of Chemistry at the solidly constituted”, having a number of University of Kazan in Russia, had dis- properties in common, that could be arranged covered the sixth member of the group and in two supcrimposed groups of thrce, the named it ruthenium in honour of his adopted “Nebenreihe” or second.ary series-ruthen- country. During the intervening period ium, rhodium and palladium-and the Claus published fourteen papers describing “Hauptreihe” or principal series-osmium, the compounds of the six metals and their iridium and platinum. Mendeliev’s revised Periodic Table, published in 1871 Platinum Metals Rev., 1960, 4, (4), 144-149 144 The PIatinum Metals and their Neighbours in the Periodic Table Sroup VIA 3oup VIIA Group VIII Group IB First long period Cr Mn Fe Co Ni cu 24 25 26 27 28 29 Second long period Mo Tc Ru Rh Pd Ag 42 43 44 45 46 47 Third long period W Re 0s Ir Pt Au 74 75 76 77 78 79 Although Dobereiner had described his opposite page that the two platinum metal triads some thirty years before this, Claus was triads described by Claus, together with the some years ahead of the famous Newland triad iron, cobalt and nickel, were placed in octaves, while the broader generalisations of Group VIII by Mendeliev, but that the Lothar Meyer and Mendeliev were still ten elements copper, silver and gold also appeared years away. -

The Discovery of Buckminsterfullerene Topics Allotropy, Organic Chemistry, Chemists
The discovery of buckminsterfullerene Topics Allotropy, organic chemistry, chemists In 1985, virtually all school chemistry textbooks became out of date overnight. Prior to this, textbooks stated that there were two allotropes of carbon – diamond and graphite. Allotropes are forms of the same element which differ in the way their atoms are arranged. Diamond and graphite are classic examples of allotropy. In diamond the carbon atoms are arranged tetrahedrally and in graphite they form two- dimensional layers of interlinked hexagons. In 1985 the discovery was announced of a third allotrope in which the atoms form C60 molecules in the shape of a football. This led to the award of the 1996 Nobel Prize to Harry (now Sir Harry) Kroto of Sussex University, Robert Curl and Richard Smalley (both of Rice University in Houston, USA). Harry Kroto had an interest in molecules found in interstellar space which can be identified from their microwave spectra – by comparing signals obtained from outer space with those measured for known compounds in the laboratory. He was particularly interested in poly-ynes (molecules with several carbon-carbon triple bonds) but these are difficult to make conventionally. In 1984, Kroto began a collaboration with Smalley and Curl in Houston who had an apparatus called AP2 (‘App-two’). This used a laser to blast clusters of atoms off solid targets and then led the clusters into a mass spectrometer where their relative molecular masses could be measured. Kroto hoped that if a graphite target were used, small sections of graphite layers might rearrange themselves into poly-ynes whose spectra could then be measured.