Computational Study of Allotropic Structures of Carbon by Density Functional Theory (DTF)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Group Vi Elements (The Chalcogens)
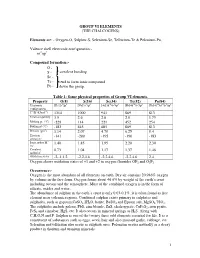
GROUP VI ELEMENTS (THE CHALCOGENS) Elements are: - Oxygen-O, Sulphur-S, Selenium-Se, Tellurium-Te & Polonium-Po. Valence shell electronic configuration:- ns2np4 Compound formation:- O - S - covalent bonding Se - Te - tend to form ionic compound Po - down the group. Table 1: Some physical properties of Group VI elements. Property O(8) S(16) Se(34) Te(52) Po(84) Electronic [He]2s22p4 [Ne]3s23p4 [Ar]3d104s24p4 [Kr]4d105s25p4 [Xe]4f145d106s26p4 configuration 1st IE (kJmol-1) 1314 1000 941 869 813 Electronegativity 3.5 2.6 2.6 2.0 1.75 Melting pt. (oC) -229 114 221 452 254 Boiling pt (oC) -183 445 685 869 813 Density (gm-3) 1.14 2.07 4.79 6.25 9.4 Electron -141 -200 -195 -190 -183 affinity,E- Ionic radius M2- 1.40 1.85 1.95 2.20 2.30 /Ao Covalent 0.73 1.04 1.17 1.37 1.46 radius/Ao Oxidation states -2,-1,1,2 -2,2,4,6 -2,2,4,6 -2,2,4,6 2,4 Oxygen shows oxidation states of +1 and +2 in oxygen fluorides OF2 and O2F2 Occurrence:- Oxygen is the most abundant of all elements on earth. Dry air contains 20.946% oxygen by volume in the free form. Oxygen forms about 46.6% by weight of the earth’s crust including oceans and the atmosphere. Most of the combined oxygen is in the form of silicate, oxides and water. The abundance of sulphur in the earth’s crust is only 0.03-0.1%. it is often found as free element near volcanic regions. -

Graphene – Diamond Nanomaterials: a Status Quo Review
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 29 July 2021 doi:10.20944/preprints202107.0647.v1 Article Graphene – diamond nanomaterials: a status quo review 1 Jana Vejpravová ,* 1 Department of Condensed Matter Physics, Faculty of Mathematics and Physics, Charles University, Ke Karlovu 5, 121 16 Prague 2, Czech Republic. * Correspondence: JV, [email protected] Abstract: Carbon nanomaterials with a different character of the chemical bond – graphene (sp2) and nanodiamond (sp3) are the building bricks for a new class of all-carbon hybrid nanomaterials, where the two different carbon networks with the sp3 and sp2 hybridization coexist, interact and even transform into one another. The unique electronic, mechanical, and chemical properties of the two border nanoallotropes of carbon ensure the immense application potential and versatility of these all-carbon graphene – diamond nanomaterials. The review gives an overview of the current state of the art of graphene – diamond nanomaterials, including their composites, heterojunctions, and other hybrids for sensing, electronic, energy storage, and other applications. Also, the graphene- to-diamond and diamond-to-graphene transformations at the nanoscale, essential for innovative fabrication, and stability and chemical reactivity assessment are discussed based on extensive theo- retical, computational, and experimental studies. Keywords: graphene; diamond; nanodiamond; diamane; graphene-diamond nanomaterials; all car- bon materials; electrochemistry; mechanochemistry; sensor; supercapacitor; -

X-Ray Topographic Investigation of Diamond Anvils for High Pressure Generation - Correlation Between Defects and Early Failure
X-ray topographic investigation of diamond anvils for high pressure generation - Correlation between defects and early failure Dewaele A. and Loubeyre P. CEA/DPTA, BP12, 91680 Bruyères le Châtel The diamond anvil cell technique revolutionized high pressure physics some 25 years ago. This device takes advantage of the unusual mechanical properties of the diamond. A metallic gasket with a hole in it to confine the sample is compressed between two diamond anvils. This device allows the generation of pressures that reach 300 GPa (3 millions times atmospheric pressure), pressure at which the diamond exhibits large elastic strain [1]. The pressure reached in diamond anvil cells is very often limited by the failure of a diamond anvil. In order to prevent this phenomenon, anvils are selected on the basis of their chemical purity, and their internal strains. However, the chemical purity does not guarantee the resistance of diamond anvils under high pressure operation. In particular, when the diamonds are used in contact with H2 or Helium samples, species which diffuse in diamond. Unfortunately, helium is known to be the best pressure transmitting medium, and is often loaded for that use in diamonds anvil cells [2]. A better understanding of early breakdown of diamond anvils and an a priori diagnostic of their resistance would thus constitute a major improvement for high pressure techniques. X-ray topography helps to establish this anvils quality diagnostic, because this method evidences intrinsic crystallographic defects of the anvils. These defects are likely to influence the mechanical properties of the anvil [3]. However, the liability of x-ray topography diagnostic had to be proven. -

Ecological Comparison of Synthetic Versus Mined Diamonds
Ecological Comparison of Synthetic versus Mined Diamonds Saleem H. Ali Working Paper, Institute for Environmental Diplomacy and Security University of Vermont, January, 2011 http://www.uvm.edu/ieds Abstract The energy usage and emissions in mined versus lab-created diamonds was evaluated, based on industrial data, since these two factors are often a general indicator of environmental impact that can be useful in product comparisons. Depending on the process and the location of the mine, the data can be highly divergent and cannot be used as a singular measure of environmental impact. There is a need to develop life cycle analysis techniques from industrial ecology to conduct a detailed comparison of synthetic versus mined stones. Introduction Synthetic diamonds have come of age, and the year 2010 will be remembered as a landmark year in this regard since for the first time labs of the Gemological Institute of America (GIA) in New York were able to grade a gem quality near-colorless synthetic diamond (formed by chemical vapor deposition) , greater than 1 carat. The history of synthetic diamonds at the industrial level, goes back to patents for super-abrasives at General Electric can be traced back to several decades (Hazen, 1996). However, gem quality synthetic diamonds have only risen to prominence in the last decade with the rise of a few key companies who are taking on this growing market in concert with boutique jewelry brands. As jewelers consider environmental social responsibility more seriously in marketing their gemstone products, energy usage in mined versus lab-created gems can be an important factor in determining comparative environmental impact. -

Basic Keyword List
NOTICE TO AUTHORS 2008 Basic Keyword list A Antibodies Bismuth Chalcogens Ab initio calculations Antifungal agents Block copolymers Chaperone proteins Absorption Antigens Bond energy Charge carrier injection Acidity Antimony Bond theory Charge transfer Actinides Antisense agents Boranes Chelates Acylation Antitumor agents Borates Chemical vapor deposition Addition to alkenes Antiviral agents Boron Chemical vapor transport Addition to carbonyl com- Aqueous-phase catalysis Bridging ligands Chemisorption pounds Arene ligands Bromine Chemoenzymatic synthesis Adsorption Arenes Brønsted acids Chemoselectivity Aerobic oxidation Argon Chiral auxiliaries Aggregation Aromatic substitution C Chiral pool Agostic interactions Aromaticity C-C activation Chiral resolution Alanes Arsenic C-C bond formation Chirality Alcohols Arylation C-C coupling Chlorine Aldehydes Aryl halides C-Cl bond activation Chromates Aldol reaction Arynes C-Glycosides Chromium Alkali metals As ligands C-H activation Chromophores Alkaline earth metals Asymmetric amplification C1 building blocks Circular dichroism Alkaloids Asymmetric catalysis Cadmium Clathrates Alkanes Asymmetric synthesis Cage compounds Clays Alkene ligands Atmospheric chemistry Calcium Cleavage reactions Alkenes Atom economy Calixarenes Cluster compounds Alkylation Atropisomerism Calorimetry Cobalamines Alkyne ligands Aurophilicity Carbanions Cobalt Alkynes Autocatalysis Carbene homologues Cofactors Alkynylation Automerization Carbene ligands Colloids Allenes Autoxidation Carbenes Combinatorial chemistry Allosterism -

OF SYNTHETIC DIAMONDS. Introduction
PI ^ AU9817130 IONOLUMINESCENCE (IL) OF SYNTHETIC DIAMONDS. A. A. Bettiol, K. W. Nugent, D. N. Jamieson and S. Prawer School of Physics, Microanalytical Research Centre, University of Melbourne, Parkville, 3052, AUSTRALIA. Introduction The optical properties of natural and synthetic diamonds have been extensively characterized in the past by absorption and luminescence. The use of such techniques as cathodoluminescence, photoluminescence, photoluminescence excitation and electron spin and paramagnetic resonance has resulted in the identification of many impurity and defect related optical centres in diamond [1-2]. Of the impurities found in diamond, nitrogen is by far the most abundant and hence responsible for most of the optical properties [3]. The development of diamond synthesis methods has resulted in the discovery of a number of a new optically active impurities and defects which are introduced during the growth process. These include Si, O, Ni and B [1-2]. In this study we identify a number of defect and impurity related centres in two commercially produced synthetic diamond samples by using the novel technique of ionoluminescence (IL) [4]. The first sample characterized is a Norton polycrystalline diamond detector. Signal produced in any charged particle detector is degraded if recombination of the electrons and holes occurs before the charge can be swept out by the electric field in the detector. In diamonds where the radiative recombination cross-section can be quite high, signal degradation can occur depending on the optical centres present and their lifetimes. Recombination centres with lifetimes much longer than the sweep out time will potentially saturate hence only cause a degradation of signal. -

DIAMOND Natural Colorless Type Iab Diamond with Silicon-Vacancy
Editors Thomas M. Moses | Shane F. McClure DIAMOND logical and spectroscopic features con- Natural Colorless Type IaB firmed the diamond’s natural origin, – Diamond with Silicon-Vacancy despite the occurrence of [Si-V] emis- Defect Center sions. No treatment was detected. Examination of this stone indicated The silicon-vacancy defect, or [Si-V]–, that the [Si-V]– defect can occur, albeit is one of the most important features rarely, in multiple types of natural dia- in identifying CVD synthetic dia- monds. Therefore, all properties should monds. It can be effectively detected be carefully examined in reaching a using laser photoluminescence tech- conclusion when [Si-V]– is present. nology to reveal sharp doublet emis- sions at 736.6 and 736.9 nm. This Carmen “Wai Kar” Lo defect is extremely rare in natural dia- monds (C.M. Breeding and W. Wang, “Occurrence of the Si-V defect center Figure 1. Emissions from the Screening of Small Yellow Melee for in natural colorless gem diamonds,” silicon-vacancy defect at 736.6 and Treatment and Synthetics Diamond and Related Materials, Vol. 736.9 nm were detected in this Diamond treatment and synthesis 17, No. 7–10, pp. 1335–1344) and has 0.40 ct type IaB natural diamond. have undergone significant develop- been detected in very few natural type ments in the last decade. During this IIa and IaAB diamonds over the past showed blue fluorescence with natural time, the trade has grown increasingly several years. diamond growth patterns. These gemo- concerned about the mixing of treated Recently, a 0.40 ct round brilliant diamond with D color and VS2 clarity (figure 1) was submitted to the Hong Figure 2. -

Black Carbon and Its Impact on Earth's Climate
Lesson Plan: Black Carbon and its Impact on Earth’s Climate A teacher-contributed lesson plan by Dr. Shefali Shukla, Sri Venkateswara College (University of Delhi), India. As a High School or Undergraduate Chemistry or Environmental Sciences teacher, you can use this set of computer-based tools to teach about allotropy, various allotropes of carbon and their structural and physical properties, black carbon, sources of black carbon and its impact on Earth’s climate. This lesson plan will help students understand the concept of allotropy and various allotropes of carbons. Students will learn about black carbon, the effect of black carbon on the Earth’s albedo and therefore, its impact on the climate. This lesson plan will also help students to understand how the immediate effect of controlling black carbon emission can potentially slow down the rate of global warming. Thus, the use of this lesson plan allows you to integrate the teaching of a climate science topic with a core topic in Chemistry or Environmental Sciences. Use this lesson plan to help your students find answers to: • What are allotropes? What are the various allotropes of carbon and their properties? • What are the sources of black carbon? • What are the different effects of black carbon on clouds? How does it modify rainfall pattern? • How does the deposition of black carbon on ice caps affect melting of the ice? • Explain how black carbon can have a cooling or warming effect on the planet? • What is the effect of black carbon on human health? About the Lesson Plan Grade Level: High School, Undergraduate Discipline: Chemistry, Environmental Sciences Topic(s) in Discipline: Allotropy, Allotropes of carbon, Black Carbon, Sources of Black Carbon, Heating and Cooling Effects of Black Carbon, Effect of Black Carbon on Human Health, Black Carbon Albedo, Black Carbon Emission Climate Topic: Climate and the Atmosphere, The Greenhouse Gas Effect, Climate and the Anthroposphere Location: Global Access: Online, Offline Language(s): English Approximate Time Required: 90-120 min 1 Contents 1. -

De Beers Natural Versus Synthetic Diamond Verification Instruments
DE BEERSNATURAL VERSUS By Christopher M.Welbourn, Martin Cooper, and Paul M,Spear Twoinstruments have been developed at he subject of cuttable-quality synthetic diamonds De Beers DTC Research Centre, has been receiving much attention in the gem trade Maidenhead, to distinguish synthetic dia- recently. Yellow to yellow-brown synthetic diamonds grown monds from natural diamonds. The in Russia have been offered for sde at a number of recent DiainondSureTMenables the rapid exami- gem and jewelry trade shows (Shor and Weldon, 1996; nation of large numbers of polished dia- monds, both loose and set in jewehy. Reinitz, 1996; Johnson and Koivula, 1996))and a small num- Automatically and with high sensitivity, ber of synthetic diamonds have been submitted to gem grad- this instrument detects the presence of the ing laboratories for identification reports (Fryer, 1987; 415 nm optical absorption line, which is Reinitz, 1996; Moses et al., 1993a and b; Emms, 1994; found in the vast majority of natural dia- Kammerling et al., 1993, 1995; Kammerling and McClure, monds but not in synthetic diamonds. 1995). Particular concern was expressed following recent Those stones in which this line is detected announcements of the production and planned marketing of are "passed" by the instrument, and those near-colorless synthetic diamonds (Koivula et al., 1994; in which it is not detected are "referredfor "Upfront," 1995). further tests." The DiamondViewTRfpro- Synthetic diamonds of cuttable size and quality, and the duces a fluorescence image of the surface of technology to produce them, are not new. In 1971, researchers a polished diamond, from which the growth structure of the stone may be deter- at the General Electric Company published the results of mined. -

Introduction to Phase Diagrams*
ASM Handbook, Volume 3, Alloy Phase Diagrams Copyright # 2016 ASM InternationalW H. Okamoto, M.E. Schlesinger and E.M. Mueller, editors All rights reserved asminternational.org Introduction to Phase Diagrams* IN MATERIALS SCIENCE, a phase is a a system with varying composition of two com- Nevertheless, phase diagrams are instrumental physically homogeneous state of matter with a ponents. While other extensive and intensive in predicting phase transformations and their given chemical composition and arrangement properties influence the phase structure, materi- resulting microstructures. True equilibrium is, of atoms. The simplest examples are the three als scientists typically hold these properties con- of course, rarely attained by metals and alloys states of matter (solid, liquid, or gas) of a pure stant for practical ease of use and interpretation. in the course of ordinary manufacture and appli- element. The solid, liquid, and gas states of a Phase diagrams are usually constructed with a cation. Rates of heating and cooling are usually pure element obviously have the same chemical constant pressure of one atmosphere. too fast, times of heat treatment too short, and composition, but each phase is obviously distinct Phase diagrams are useful graphical representa- phase changes too sluggish for the ultimate equi- physically due to differences in the bonding and tions that show the phases in equilibrium present librium state to be reached. However, any change arrangement of atoms. in the system at various specified compositions, that does occur must constitute an adjustment Some pure elements (such as iron and tita- temperatures, and pressures. It should be recog- toward equilibrium. Hence, the direction of nium) are also allotropic, which means that the nized that phase diagrams represent equilibrium change can be ascertained from the phase dia- crystal structure of the solid phase changes with conditions for an alloy, which means that very gram, and a wealth of experience is available to temperature and pressure. -

Large Man-Made Diamond Single Crystals: Manufacture, Properties and Application
Large man-made diamond single crystals: manufacture, properties and application Dr. Sergey A.Terentjev , the head of Department of Single Crystal Growth. Technological Institute for Superhard and Novel Carbon Materials ( ТISN СМ ) E-mail: Diamond is a material possessing a unique complex of physical and chemical properties which potentially allow using it, besides traditional application in the processing industry, in Hi- Tech branches also. Such use of diamond restrains by insufficient quantity of natural perfect raw material of so-called "device" quality. Single crystals of diamond grown up in laboratory conditions differ from natural, first of all, in high reproducibility of physical properties (impurities level, an optical transparency, heat conductivity, specific electric resistance, etc.). Creation of technologies for cultivation a high purity «nitrogen free» (IIa type) and semi- conductor (IIb type) large diamond single crystals, which are extremely rare among natural diamonds and very expensive, is of the great interest and simultaneously of the greatest difficulty. Technologies for manufacture of both IIa and IIb diamond single crystals 0.5 to 5.0 carat in weight (characteristic size 3÷8 mm) from solvent/catalyst melt of transitive metals at high static pressures and temperatures (based on a temperature gradient growth technique) have been developed by Dr. Terentiev’s research team some years ago and successfully introduced in industrial department of ТISN СМ . Man-made IIa type single crystals are characterized by an optical transparency in a less than 25 µm wave lengths region and have fundamental absorption edge at 225 nanometers. Their thermal conductivity reaches 2000 Wt/m• К. Nitrogen concentration (a basic impurity element) level does not exceed 2 рр m (usually less than 1 рр m). -

Method of Making Synthetic Diamond Film
Europaisches Patentamt European Patent Office © Publication number: 0 597 445 A2 Office europeen des brevets EUROPEAN PATENT APPLICATION © Application number: 93118150.7 int. Ci.5; C23C 16/00, C23C 16/26 @ Date of filing: 09.11.93 ® Priority: 10.11.92 US 973994 32 Cornell Street Arlington, MA 02174(US) @ Date of publication of application: Inventor: Frey, Robert M. 18.05.94 Bulletin 94/20 77 Santa Isabel Nr.2-14 Laguna Vista, Texas 78578(US) © Designated Contracting States: DE FR GB 0 Representative: Diehl, Hermann, Dr. © Applicant: NORTON COMPANY Dipl.-Phys. et al 1 New Bond Street DIEHL, GLASER, HILTL & PARTNER Worcester, MA 01615-0008(US) Patentanwalte Fluggenstrasse 13 @ Inventor: Simpson, Matthew D-80639 Munchen (DE) © Method of making synthetic diamond film. © The invention describes a method for making a free-standing synthetic diamond film of desired thickness, comprising the steps of: no SELECT TARGET providing a substrate; X THICKNESS OF DIAMOND selecting a target thickness of diamond to be TO BE PRODUCED produced, said target thickness being in the range 200 urn to 1000 urn; finishing a surface of the substrate to a rough- 1 20 finish surface of ness, RA that is a function of the target thickness, substrate to prescribed said roughness being determined from ra Roughness 0.38t/600 urn ^ Ra = 0.50 urn 200 urn < t ^ 600 urn 0.38 urn ^ Ra = 0.50 urn 600 urn < t < 1000 1 30, urn DEPOSIT INTERLAYER where t is the target thickness; said the inter- CM depositing an interlayer on substrate, layer having a thickness in the range 1 to 20 urn; depositing synthetic diamond on said interlayer, by I40.