Pnictogens Allotropy and Phase Transformation During Van Der Waals Growth
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metastability of Anatase
Metastability of anatase: size dependent and irreversible anatase-rutile phase SUBJECT AREAS: DENDRIMERS transition in atomic-level precise titania POLYMER CHEMISTRY Norifusa Satoh1, Toshio Nakashima2 & Kimihisa Yamamoto2 NANOSCIENCE AND TECHNOLOGY COORDINATION CHEMISTRY 1Photovoltaic Materials Unit, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki, 305-0047 Japan, 2Chemical Resources Laboratory, Tokyo Institute of Technology, 4259 Nagatsuta, Yokohama 226-8503 Japan. Received 12 March 2013 Since crystal phase dominantly affects the properties of nanocrystals, phase control is important for the applications. To demonstrate the size dependence in anatase-rutile phase transition of titania, we used Accepted quantum-size titania prepared from the restricted number of titanium ions within dendrimer templates for 21 May 2013 size precision purposes and optical wave guide spectroscopy for the detection. Contrary to some theoretical calculations, the observed irreversibility in the transition indicates the metastablity of anatase; Published thermodynamics cannot explain the formation of metastable states. Therefore, we take into account the 7 June 2013 kinetic control polymerization of TiO6 octahedral units to explain how the crystal phase of the crystal-nucleus-size titania is dependent on which coordination sites, cis-ortrans-, react in the TiO6 octahedra, suggesting possibilities for the synthetic phase control of nanocrystals. In short, the dendrimer Correspondence and templates give access to crystal nucleation chemistry. The paper will also contribute to the creation of artificial metastable nanostructures with atomic-level precision. requests for materials should be addressed to N.S. (SATOH. he size dependence of nanocrystals during a solid-solid phase transition has attracted much attention1,2, [email protected]) because phase control is a key step to improve functionalities in photovoltaics3,4, ferroelectricity5, magnetics6, 7 or K.Y. -
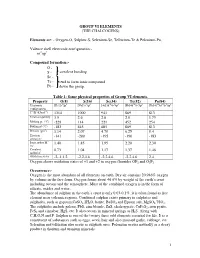
Group Vi Elements (The Chalcogens)
GROUP VI ELEMENTS (THE CHALCOGENS) Elements are: - Oxygen-O, Sulphur-S, Selenium-Se, Tellurium-Te & Polonium-Po. Valence shell electronic configuration:- ns2np4 Compound formation:- O - S - covalent bonding Se - Te - tend to form ionic compound Po - down the group. Table 1: Some physical properties of Group VI elements. Property O(8) S(16) Se(34) Te(52) Po(84) Electronic [He]2s22p4 [Ne]3s23p4 [Ar]3d104s24p4 [Kr]4d105s25p4 [Xe]4f145d106s26p4 configuration 1st IE (kJmol-1) 1314 1000 941 869 813 Electronegativity 3.5 2.6 2.6 2.0 1.75 Melting pt. (oC) -229 114 221 452 254 Boiling pt (oC) -183 445 685 869 813 Density (gm-3) 1.14 2.07 4.79 6.25 9.4 Electron -141 -200 -195 -190 -183 affinity,E- Ionic radius M2- 1.40 1.85 1.95 2.20 2.30 /Ao Covalent 0.73 1.04 1.17 1.37 1.46 radius/Ao Oxidation states -2,-1,1,2 -2,2,4,6 -2,2,4,6 -2,2,4,6 2,4 Oxygen shows oxidation states of +1 and +2 in oxygen fluorides OF2 and O2F2 Occurrence:- Oxygen is the most abundant of all elements on earth. Dry air contains 20.946% oxygen by volume in the free form. Oxygen forms about 46.6% by weight of the earth’s crust including oceans and the atmosphere. Most of the combined oxygen is in the form of silicate, oxides and water. The abundance of sulphur in the earth’s crust is only 0.03-0.1%. it is often found as free element near volcanic regions. -

Multiple Interfaces in Diffusional Phase Transitions in Binary Mesogen-Non-Mesogen Mixtures Undergoing Metastable Phase Separations
Multiple interfaces in diffusional phase transitions in binary mesogen-non-mesogen mixtures undergoing metastable phase separations Ezequiel R. Soulé 1,2 , Cyrille Lavigne 2,3 , Linda Reven 3 and Alejandro D. Rey 2* 1. Institute of Materials Science and Technology (INTEMA), University of Mar del Plata and National Research Council (CONICET), J. B. Justo 4302, 7600 Mar del Plata, Argentina 2. Department of Chemical Engineering, McGill University, Montreal, Quebec H3A 2B2, Canada 3. Center for Self-Assembled Chemical Structure (CSACS), Chemistry Department, McGill University, 801 Sherbrooke St. West, Montreal, Quebec H3A 2K6, Canada * corresponding author Abstract Theory and simulations of simultaneous chemical demixing and phase ordering are performed for a mixed order parameter system with an isotropic-isotropic (I-I) phase separation that is metastable with respect to an isotropic-nematic (I-N) phase ordering transition. Under certain conditions, the disordered phase transforms into an ordered phase via the motion of a double front containing a metastable phase produced by I-I demixing, a thermodynamically driven mechanism not previously reported. Different kinetic regimes are found depending on the location of the initial conditions in the thermodynamic phase diagram and the ratio between diffusional and nematic phase ordering mobilities. For a diffusional process, depending if the temperature is above or below the critical co-dissolution point, an inflection point or a phase separation takes place in the depletion layer. This phase separation leads to the formation of a second interface where the separation of the two metastable isotropic phases grows monotonically with time. The observed deviations from the typical Fickian concentration profiles are associated with strong positive deviations of the mixture from ideality due to couplings between concentration and nematic ordering. -

Reactions of Lithium Nitride with Some Unsaturated Organic Compounds. Perry S
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1963 Reactions of Lithium Nitride With Some Unsaturated Organic Compounds. Perry S. Mason Jr Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Mason, Perry S. Jr, "Reactions of Lithium Nitride With Some Unsaturated Organic Compounds." (1963). LSU Historical Dissertations and Theses. 898. https://digitalcommons.lsu.edu/gradschool_disstheses/898 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. This dissertation has been 64—5058 microfilmed exactly as received MASON, Jr., Perry S., 1938- REACTIONS OF LITHIUM NITRIDE WITH SOME UNSATURATED ORGANIC COMPOUNDS. Louisiana State University, Ph.D., 1963 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. REACTIONS OF LITHIUM NITRIDE WITH SOME UNSATURATED ORGANIC COMPOUNDS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requireiaents for the degree of Doctor of Philosophy in The Department of Chemistry by Perry S. Mason, Jr. B. S., Harding College, 1959 August, 1963 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. -

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -

High-Quality, Low-Cost Bulk Gallium Nitride Substrates
ADVANCED MANUFACTURING OFFICE High-Quality, Low- Cost Bulk Gallium Nitride Substrates Electrochemical Solution Growth: A Scalable Semiconductor Manufacturing Process The ever-growing demand in the past decade for more energy efficient solid-state lighting and electrical power conversion is leading to a higher demand for wide bandgap semiconductor-based devices, such as gallium nitride (GaN), over traditional silicon (Si)-based devices. High cost and limited availability, how- ever, have hindered the adoption of GaN substrates to date. When utilizing GaN, current LED and power electronic device applications employ GaN epitaxially grown on top of non-GaN substrates. The lattice mismatch between the epitaxial GaN layer and the non-native substrate surface leads to con- siderable stress and high defect densities, ultimately compromising device yield and Conceptual diagram of the ESG reactor. Photo courtesy of Sandia National Laboratories performance. While bulk growth of GaN can combat these issues, current growth methods for bulk GaN have not fostered widespread adoption to date due to lim- This project will help develop ESG into a viable GaN bulk growth process that is well ited scalability, low material quality, high suited for scalability to large-area wafer manufacturing. Bulk GaN is important to bol- operating temperatures and pressures, stering U.S. competitiveness in high-efficiency power electronics and solid-state lighting. and slow growth rates. A fundamentally different manufacturing route for bulk Benefits for Our Industry and Our Nation growth of GaN not driven by thermal pro- Scaling the ESG growth method to large area GaN crystals could reduce the production cesses is needed to provide an adequate cost of bulk GaN wafers by up to a factor of 10. -

Basic Keyword List
NOTICE TO AUTHORS 2008 Basic Keyword list A Antibodies Bismuth Chalcogens Ab initio calculations Antifungal agents Block copolymers Chaperone proteins Absorption Antigens Bond energy Charge carrier injection Acidity Antimony Bond theory Charge transfer Actinides Antisense agents Boranes Chelates Acylation Antitumor agents Borates Chemical vapor deposition Addition to alkenes Antiviral agents Boron Chemical vapor transport Addition to carbonyl com- Aqueous-phase catalysis Bridging ligands Chemisorption pounds Arene ligands Bromine Chemoenzymatic synthesis Adsorption Arenes Brønsted acids Chemoselectivity Aerobic oxidation Argon Chiral auxiliaries Aggregation Aromatic substitution C Chiral pool Agostic interactions Aromaticity C-C activation Chiral resolution Alanes Arsenic C-C bond formation Chirality Alcohols Arylation C-C coupling Chlorine Aldehydes Aryl halides C-Cl bond activation Chromates Aldol reaction Arynes C-Glycosides Chromium Alkali metals As ligands C-H activation Chromophores Alkaline earth metals Asymmetric amplification C1 building blocks Circular dichroism Alkaloids Asymmetric catalysis Cadmium Clathrates Alkanes Asymmetric synthesis Cage compounds Clays Alkene ligands Atmospheric chemistry Calcium Cleavage reactions Alkenes Atom economy Calixarenes Cluster compounds Alkylation Atropisomerism Calorimetry Cobalamines Alkyne ligands Aurophilicity Carbanions Cobalt Alkynes Autocatalysis Carbene homologues Cofactors Alkynylation Automerization Carbene ligands Colloids Allenes Autoxidation Carbenes Combinatorial chemistry Allosterism -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

Black Carbon and Its Impact on Earth's Climate
Lesson Plan: Black Carbon and its Impact on Earth’s Climate A teacher-contributed lesson plan by Dr. Shefali Shukla, Sri Venkateswara College (University of Delhi), India. As a High School or Undergraduate Chemistry or Environmental Sciences teacher, you can use this set of computer-based tools to teach about allotropy, various allotropes of carbon and their structural and physical properties, black carbon, sources of black carbon and its impact on Earth’s climate. This lesson plan will help students understand the concept of allotropy and various allotropes of carbons. Students will learn about black carbon, the effect of black carbon on the Earth’s albedo and therefore, its impact on the climate. This lesson plan will also help students to understand how the immediate effect of controlling black carbon emission can potentially slow down the rate of global warming. Thus, the use of this lesson plan allows you to integrate the teaching of a climate science topic with a core topic in Chemistry or Environmental Sciences. Use this lesson plan to help your students find answers to: • What are allotropes? What are the various allotropes of carbon and their properties? • What are the sources of black carbon? • What are the different effects of black carbon on clouds? How does it modify rainfall pattern? • How does the deposition of black carbon on ice caps affect melting of the ice? • Explain how black carbon can have a cooling or warming effect on the planet? • What is the effect of black carbon on human health? About the Lesson Plan Grade Level: High School, Undergraduate Discipline: Chemistry, Environmental Sciences Topic(s) in Discipline: Allotropy, Allotropes of carbon, Black Carbon, Sources of Black Carbon, Heating and Cooling Effects of Black Carbon, Effect of Black Carbon on Human Health, Black Carbon Albedo, Black Carbon Emission Climate Topic: Climate and the Atmosphere, The Greenhouse Gas Effect, Climate and the Anthroposphere Location: Global Access: Online, Offline Language(s): English Approximate Time Required: 90-120 min 1 Contents 1. -

Naming Compounds Practice Problems KEY Naming Simple Ionic Compounds
Chemistry HS/Science Unit: 05 Lesson: 01 Naming Compounds Practice Problems KEY Naming Simple Ionic Compounds Name the following compounds: 1) KCl potassium chloride 2) MgI2 magnesium iodide 3) FeO iron (II) oxide 4) Fe2O3 iron (III) oxide 5) Cu3P copper (I) phosphide 6) SnSe2 tin (IV) selenide 7) TiBr3 titanium (III) bromide 8) GaAs gallium arsenide 9) BeF2 beryllium fluoride 10) Cs3N cesium nitride Write the formulas for the following compounds: 1) lithium iodide LiI 2) cobalt (III) oxide Co2O3 3) calcium fluoride CaF2 4) silver bromide AgBr 5) sodium hydride NaH 6) vanadium (V) sulfide V2S5 7) lead (II) nitride Pb3N2 8) titanium (II) selenide TiSe 9) manganese (VII) arsenide Mn3As7 10) gallium chloride GaCl3 ©2013, TESCCC 06/17/13 page 1 of 4 Chemistry HS/Science Unit: 05 Lesson: 01 Naming Compounds Practice Problems Naming Complex (polyatomic) Ionic Compounds Name the following compounds: 1) NH4Cl ammonium chloride 2) Fe(NO3)3 iron (III) nitrate 3) Pb(SO4)2 lead (IV) sulfate 4) Ag3PO4 silver phosphate 5) Be(HCO3)2 beryllium hydrogen carbonate 6) Al(CN)3 aluminum cyanide 7) Mn2(SO3)3 manganese (III) sulfite 8) Sr(C2H3O2)2 strontium acetate 9) Ti(CN)4 titanium (IV) cyanide 10) YClO3 yttrium chlorate Write the formulas for the following compounds: 1) lead (IV) sulfate Pb(SO4)2 2) silver cyanide AgCN 3) copper (II) chlorate Cu(ClO3)2 4) chromium (IV) phosphate Cr3(PO4)4 5) vanadium (IV) carbonate V(CO3)2 6) ammonium oxide (NH4)2O 7) tin (II) nitrite Sn(NO2)2 8) chromium (III) hydroxide Cr(OH)3 9) titanium (II) acetate Ti(C2H3O2)2 10) -

Introduction to Phase Diagrams*
ASM Handbook, Volume 3, Alloy Phase Diagrams Copyright # 2016 ASM InternationalW H. Okamoto, M.E. Schlesinger and E.M. Mueller, editors All rights reserved asminternational.org Introduction to Phase Diagrams* IN MATERIALS SCIENCE, a phase is a a system with varying composition of two com- Nevertheless, phase diagrams are instrumental physically homogeneous state of matter with a ponents. While other extensive and intensive in predicting phase transformations and their given chemical composition and arrangement properties influence the phase structure, materi- resulting microstructures. True equilibrium is, of atoms. The simplest examples are the three als scientists typically hold these properties con- of course, rarely attained by metals and alloys states of matter (solid, liquid, or gas) of a pure stant for practical ease of use and interpretation. in the course of ordinary manufacture and appli- element. The solid, liquid, and gas states of a Phase diagrams are usually constructed with a cation. Rates of heating and cooling are usually pure element obviously have the same chemical constant pressure of one atmosphere. too fast, times of heat treatment too short, and composition, but each phase is obviously distinct Phase diagrams are useful graphical representa- phase changes too sluggish for the ultimate equi- physically due to differences in the bonding and tions that show the phases in equilibrium present librium state to be reached. However, any change arrangement of atoms. in the system at various specified compositions, that does occur must constitute an adjustment Some pure elements (such as iron and tita- temperatures, and pressures. It should be recog- toward equilibrium. Hence, the direction of nium) are also allotropic, which means that the nized that phase diagrams represent equilibrium change can be ascertained from the phase dia- crystal structure of the solid phase changes with conditions for an alloy, which means that very gram, and a wealth of experience is available to temperature and pressure. -

Boron-Nitride-Datasheet-V1
A Unique Proposition for Industry The introduction of boron nitride into our stable of 2D materials widens the focus of delivering innovative solutions for industry Neill Ricketts, CEO Our latest 2D success story Hexotene is a few-layer hexagonal boron nitride (h-BN) nanoplatelet powder with large lateral dimensions. With high chemical purity and mono-layer particles confirmed, Hexotene is the latest addition to our high performance 2D product range. It’s unique characteristics, specifically with regards to electrical conductivity, show some markedly different properties when compared to graphene. This is particularly promising for combined projects using both graphene and boron nitride. Example BN Nano platelets Information Property Measurement Method Boron 42 è 2.0At.% Predominantly few-layer with some Raman Nitrogen 45 è 2.0At.% Layers mono-layer and bi-layer spectroscopy Oxygen 3.0 è 1.0At.% Lateral Carbon 8.0 è 2.0At.% up to 5.0 µm SEM dimensions Method XPS IR UV Wavelenght (nm) 4096 1024 256 Band Gap ~6eV Energy BORON NITRIDE GRAPHENE Graphene Metals Semi- Plastics Boron Conductor Nitride 0.125 0.25 0.50 1.00 2.00 4.00 8.00 Conductors Semi-Conductors Insulators Energy (eV) Diagram showing extremes of electrical conductivity, with Hexagonal boron nitride is white in colour and is unusual as boron nitride having an ultra-wide bandgap. it absorbs high energy UV light, whereas graphene absorbs all light frequencies. What is h-BN? Boron and nitrogen are neighbours of carbon in boron nitride (h-BN). It also happens to be the the Periodic Table. Just as carbon can exist as softest of the BN polymorphs.