Transcriptomic Profiling Identifies Novel Mechanisms of Transcriptional
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
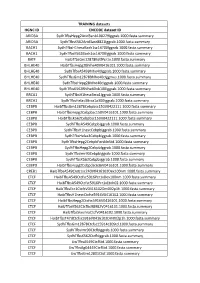
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

(12) Patent Application Publication (10) Pub. No.: US 2006/0068395 A1 Wood Et Al
US 2006.0068395A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0068395 A1 Wood et al. (43) Pub. Date: Mar. 30, 2006 (54) SYNTHETIC NUCLEIC ACID MOLECULE (21) Appl. No.: 10/943,508 COMPOSITIONS AND METHODS OF PREPARATION (22) Filed: Sep. 17, 2004 (76) Inventors: Keith V. Wood, Mt. Horeb, WI (US); Publication Classification Monika G. Wood, Mt. Horeb, WI (US); Brian Almond, Fitchburg, WI (51) Int. Cl. (US); Aileen Paguio, Madison, WI CI2O I/68 (2006.01) (US); Frank Fan, Madison, WI (US) C7H 2L/04 (2006.01) (52) U.S. Cl. ........................... 435/6: 435/320.1; 536/23.1 Correspondence Address: SCHWEGMAN, LUNDBERG, WOESSNER & (57) ABSTRACT KLUTH 1600 TCF TOWER A method to prepare synthetic nucleic acid molecules having 121 SOUTHEIGHT STREET reduced inappropriate or unintended transcriptional charac MINNEAPOLIS, MN 55402 (US) teristics when expressed in a particular host cell. Patent Application Publication Mar. 30, 2006 Sheet 1 of 2 US 2006/0068395 A1 Figure 1 Amino Acid Codon Phe UUU, UUC Ser UCU, UCC, UCA, UCG, AGU, AGC Tyr UAU, UAC Cys UGU, UGC Leu UUA, UUG, CUU, CUC, CUA, CUG Trp UGG Pro CCU, CCC, CCA, CCG His CAU, CAC Arg CGU, CGC, CGA, CGG, AGA, AGG Gln CAA, CAG Ile AUU, AUC, AUA Thr ACU, ACC, ACA, ACG ASn AAU, AAC LyS AAA, AAG Met AUG Val GUU, GUC, GUA, GUG Ala GCU, GCC, GCA, GCG Asp GAU, GAC Gly GGU, GGC, GGA, GGG Glu GAA, GAG Patent Application Publication Mar. 30, 2006 Sheet 2 of 2 US 2006/0068395 A1 Spd Sequence pGL4B-4NN3. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

JASPAR 2014: an Extensively Expanded and Updated Open-Access Database of Transcription Factor Binding Profiles
JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Anthony Mathelier, Xiaobei Zhao, Allen W Zhang, François Parcy, Rebecca Worsley-Hunt, David J Arenillas, Sorana Buchman, Chih-Yu Chen, Alice Chou, Hans Ienasescu, et al. To cite this version: Anthony Mathelier, Xiaobei Zhao, Allen W Zhang, François Parcy, Rebecca Worsley-Hunt, et al.. JASPAR 2014: an extensively expanded and updated open-access database of transcription fac- tor binding profiles.. Nucleic Acids Research, Oxford University Press, 2014, 42 (1), pp.D142-7. 10.1093/nar/gkt997. hal-00943558 HAL Id: hal-00943558 https://hal.archives-ouvertes.fr/hal-00943558 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial| 4.0 International License D142–D147 Nucleic Acids Research, 2014, Vol. 42, Database issue Published online 4 November 2013 doi:10.1093/nar/gkt997 JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles Anthony Mathelier1, Xiaobei Zhao2,3, Allen W. Zhang1, Franc¸ ois Parcy4, Rebecca Worsley-Hunt1, David J. Arenillas1, Sorana Buchman2, Chih-yu Chen1, Alice Chou1, Hans Ienasescu2, Jonathan Lim1, Casper Shyr1, Ge Tan4, Michelle Zhou1, Boris Lenhard5,6,*, Albin Sandelin2,* and Wyeth W. -

Supplemental Table 1: Snps Genotyped for NCO, Listed Alphabetically by Gene Name
Supplemental Table 1: SNPs genotyped for NCO, listed alphabetically by gene name. Gene Name SNP rs# ACVR1 rs10497189 ACVR1 rs10497191 ACVR1 rs10497192 ACVR1 rs10933441 ACVR1 rs1146035 ACVR1 rs1220110 ACVR1 rs17182166 ACVR1 rs17798043 ACVR1 rs4380178 ACVR1 rs6719924 ACVR2 rs1128919 ACVR2 rs10497025 ACVR2 rs1424941 ACVR2 rs1424954 ACVR2 rs17742573 ACVR2 rs2382112 ACVR2 rs4419186 AKT2 rs11671439 AKT2 rs12460555 AKT2 rs16974157 AKT2 rs2304188 AKT2 rs3730050 AKT2 rs7250897 AKT2 rs7254617 AKT2 rs874269 ALOX12B/ALOXE3 rs3809881 ALOX15B rs4792147 ALOX15B rs9898751 AMH rs10407022 AMH rs2074860 AMH rs3746158 AMH rs4806834 AMH rs757595 AMH rs886363 AMHR2 rs10876451 AMHR2 rs11170547 AMHR2 rs11170558 APC rs2229992 APC rs351771 APC rs41115 APC rs42427 APC rs459552 APC rs465899 APEX1 rs2275007 APEX1 rs3136820 15 APEX1 rs938883 APEX1 rs11160711 APEX1 rs1713459 APEX1 rs1713460 APEX1 rs1760941 APEX1 rs2275008 APEX1 rs3120073 APEX1 rs4465523 AR rs12011793 AR rs1204038 AR rs1337080 AR rs1337082 AR rs2207040 AR rs2361634 AR rs5031002 AR rs6152 AR rs962458 ATM rs1800058 ATM rs1800889 ATM rs11212570 ATM rs17503908 ATM rs227060 ATM rs228606 ATM rs3092991 ATM rs4987876 ATM rs4987886 ATM rs4987923 ATM rs4988023 ATM rs611646 ATM rs639923 ATM rs672655 ATR rs2229032 ATR rs10804682 ATR rs11920625 ATR rs13065800 ATR rs13085998 ATR rs13091637 ATR rs1802904 ATR rs6805118 ATR rs9856772 BACH1 rs388707 BACH1 rs1153276 BACH1 rs1153280 BACH1 rs1153284 BACH1 rs1153285 BACH1 rs17743655 BACH1 rs17744121 BACH1 rs2300301 BACH1 rs2832283 16 BACH1 rs411697 BACH1 rs425989 BARD1 rs1048108 -

Rxrα Represses NRF2-Mediated Transcription 1
Author Manuscript Published OnlineFirst on April 23, 2013; DOI: 10.1158/0008-5472.CAN-12-3386 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. RXRα represses NRF2-mediated transcription Title: RXRα Inhibits the NRF2-ARE Signalling Pathway Through A Direct Interaction With the Neh7 domain of NRF2 Running title: RXRα represses NRF2-mediated transcription 1,2 2 1 2 1 1 Hongyan Wang , Kaihua Liu , Miao Geng , Peng Gao , Xiaoyuan Wu , Yan Hai , Yangxia Li1, Yulong Li1, Lin Luo2, John D. Hayes3, Xiu Jun Wang2† and Xiuwen Tang1† 1Department of Biochemistry and Genetics, 2Department of Pharmacology, School of Medicine, Zhejiang University, Hangzhou, PR China, 310058, 3Division of Cancer Research, Medical Research Institute, Ninewells Hospital & Medical School, University of Dundee, Dundee DD1 9SY, Scotland, United Kingdom Key words: RXRα, repressor, NRF2, ARE, drug resistance †To whom correspondence should be addressed: PO BOX 18, The School of Medicine, Zhejiang University, Hangzhou, PR China 310058. Tel: +0086-571- 88208266; Fax: +0086-571-88208266; E-mail: [email protected] (Xiu Jun Wang) or [email protected] (Xiuwen Tang) Word count: 4993 Total number of figures and tables: 6 The authors disclose no potential conflicts of interest. Abbreviations: AKR1C, aldo-keto reductases 1C1 and 1C2; ARE, antioxidant response element; ATF3, activating transcription factor 3; BHA, butylated hydroxyanisole; CAR, constitutive androstane receptor; CBP, CREB, cAMP response element binding protein, binding -

Cannabidiol Induces Antioxidant Pathways in Keratinocytes By
University of Dundee Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1 Casares Perez, Laura; García, Víctor; Garrido-Rodríguez, Martín; Millán, Estrella; Collado, Juan A.; García-Martín, Adela Published in: Redox Biology DOI: 10.1016/j.redox.2019.101321 Publication date: 2020 Licence: CC BY-NC-ND Document Version Publisher's PDF, also known as Version of record Link to publication in Discovery Research Portal Citation for published version (APA): Casares Perez, L., García, V., Garrido-Rodríguez, M., Millán, E., Collado, J. A., García-Martín, A., Peñarando, J., Calzado, M. A., de la Vega, L., & Muñoz, E. (2020). Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1. Redox Biology, 28, 1-15. [101321]. https://doi.org/10.1016/j.redox.2019.101321 General rights Copyright and moral rights for the publications made accessible in Discovery Research Portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from Discovery Research Portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 Redox Biology 28 (2020) 101321 Contents lists available at ScienceDirect Redox Biology journal homepage: www.elsevier.com/locate/redox Cannabidiol induces antioxidant pathways in keratinocytes by targeting BACH1 T Laura Casaresa,1, Víctor Garcíab,c,1, Martín Garrido-Rodríguezb,c, Estrella Millánb, Juan A. -

Analysis of Gene Expression in Wild Type and Notch1 Mutant Retinal Cells by Single Cell Profiling Karolina Mizeracka1, Jeffrey M
Research Article Developmental Dynamics DOI 10.1002/dvdy.24006 Analysis of gene expression in wild type and Notch1 mutant retinal cells by single cell profiling Karolina Mizeracka1, Jeffrey M. Trimarchi1,2, Michael B. Stadler3, Constance L. Cepko1,4 1 Department of Genetics, Department of Ophthalmology, Harvard Medical School, Boston, MA 02115 2 Current Address: Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014 3 Friedrich Miescher Institute for Biomedical Research, 4058 Basel, Switzerland 4 Howard Hughes Medical Institute, Department of Genetics, Department of Ophthalmology, Harvard Medical School, Boston, MA 02115 Correspondence: Constance Cepko 77 Avenue Louis Pasteur, Boston, MA 02115 Phone: (617) 432-7618 Fax: (617) 432-7595 Running title: Single cell profiling of Notch1 mutant retinal cells Key words: retina, progenitor, microarray, cell fate Summary: • Profiling of individual Notch1 deficient and wild type postnatal retinal cells on microarrays reveals changes in gene expression obscured by whole tissue analysis • Notch1 deficient cells downregulate progenitor and cell cycle markers with a concomitant upregulation in early rod photoreceptor markers • Based on classification, single Notch1 deficient and wild type cells represent Developmental Dynamics transition from progenitor to postmitotic cell • Individual wild type retinal cells express cell type markers of both photoreceptors and interneurons Grant sponsor and number: National Institutes of Health Grant R01EY09676 Accepted Articles are accepted, unedited articles for future issues, temporarily published online in advance of the final edited version. © 2013 Wiley Periodicals, Inc. Received: Mar 04, 2013; Revised: May 02, 2013; Accepted: May 13, 2013 Developmental Dynamics Page 2 of 66 Abstract Background: The vertebrate retina comprises sensory neurons, the photoreceptors, as well as many other types of neurons and one type of glial cell. -

Supplemental Figure 1. Protein-Protein Interaction Network with Increased Expression in Fteb During the Luteal Phase
Supplemental Figure 1. Protein-protein interaction network with increased expression in FTEb during the luteal phase. Supplemental Figure 2. Protein-protein interaction network with decreased expression in FTEb during luteal phase. LEGENDS TO SUPPLEMENTAL FIGURES Supplemental Figure 1. Protein-protein interaction network with increased expression in FTEb during the luteal phase. Submission of probe sets differentially expressed in the FTEb specimens that clustered with SerCa as well as those specifically altered in FTEb luteal samples to the online I2D database revealed overlapping networks of proteins with increased expression in the four FTEb samples and/or FTEb luteal samples overall. Proteins are represented by nodes, and known and predicted first-degree interactions are represented by solid lines. Genes encoding proteins shown as large ovals highlighted in blue were exclusively found in the first comparison (Manuscript Figure 2), whereas those highlighted in red were only found in the second comparison (Manuscript Figure 3). Genes encoding proteins shown as large ovals highlighted in black were found in both comparisons. The color of each node indicates the ontology of the corresponding protein as determined by the Online Predicted Human Interaction Database (OPHID) link with the NAViGaTOR software. Supplemental Figure 2. Protein-protein interaction network with decreased expression in FTEb during the luteal phase. Submission of probe sets differentially expressed in the FTEb specimens that clustered with SerCa as well as those specifically altered in FTEb luteal samples to the online I2D database revealed overlapping networks of proteins with decreased expression in the four FTEb samples and/or FTEb luteal samples overall. Proteins are represented by nodes, and known and predicted first-degree interactions are represented by solid lines. -

Electronic Supplementary Material (ESI) for Metallomics
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2018 Table S2. Families of transcription factors involved in stress response based on Matrix Family Library Version 11.0 from MatInspector program analyzed in this work. FAMILY FAMILY INFORMATION MATRIX NAME INFORMATION F$ASG1 Activator of stress genes F$ASG1 .01 Fungal zinc cluster transcription factor Asg1 F$CIN5.01 bZIP transcriptional factor of the yAP-1 family that mediates pleiotropic drug resistance and salt tolerance F$CST6.01 Chromosome stability, bZIP transcription factor of the ATF/CREB family (ACA2) F$HAC1.01 bZIP transcription factor (ATF/CREB1 homolog) that regulates the unfolded protein response F$BZIP Fungal basic leucine zipper family F$HAC1.02 bZIP transcription factor (ATF/CREB1 homolog) that regulates the unfolded protein response F$YAP1.01 Yeast activator protein of the basic leucine zipper (bZIP) family F$YAP1.02 Yeast activator protein of the basic leucine zipper (bZIP) family F$MREF Metal regulatory element factors F$CUSE.01 Copper-signaling element, AMT1/ACE1 recognition sequence F$SKN7 Skn7 response regulator of S. cerevisiae F$SKN7.01 SKN7, a transcription factor contributing to the oxidative stress response F$XBP1.01 S.cerevisae XhoI site-binding protein I, stressinduced expression F$SXBP S.cerevisiae, XhoI site-binding protein I F$XBP1.02 Stress-induced transcriptional repressor F$HSF.01 Heat shock factor (yeast) F$HSF1.01 Trimeric heat shock transcription factor F$YHSF Yeast heat shock factors F$HSF1.02 Trimeric heat shock transcription factor F$MGA1.01 Heat shock transcription factor Mga1 F$YNIT Asperg./Neurospora-activ. -

Discerning the Role of Foxa1 in Mammary Gland
DISCERNING THE ROLE OF FOXA1 IN MAMMARY GLAND DEVELOPMENT AND BREAST CANCER by GINA MARIE BERNARDO Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Dissertation Adviser: Dr. Ruth A. Keri Department of Pharmacology CASE WESTERN RESERVE UNIVERSITY January, 2012 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Gina M. Bernardo ______________________________________________________ Ph.D. candidate for the ________________________________degree *. Monica Montano, Ph.D. (signed)_______________________________________________ (chair of the committee) Richard Hanson, Ph.D. ________________________________________________ Mark Jackson, Ph.D. ________________________________________________ Noa Noy, Ph.D. ________________________________________________ Ruth Keri, Ph.D. ________________________________________________ ________________________________________________ July 29, 2011 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. DEDICATION To my parents, I will forever be indebted. iii TABLE OF CONTENTS Signature Page ii Dedication iii Table of Contents iv List of Tables vii List of Figures ix Acknowledgements xi List of Abbreviations xiii Abstract 1 Chapter 1 Introduction 3 1.1 The FOXA family of transcription factors 3 1.2 The nuclear receptor superfamily 6 1.2.1 The androgen receptor 1.2.2 The estrogen receptor 1.3 FOXA1 in development 13 1.3.1 Pancreas and Kidney