Discerning the Role of Foxa1 in Mammary Gland
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

AGR2, an Endoplasmic Reticulum Protein, Is Secreted Into the Gastrointestinal Mucus
AGR2, an Endoplasmic Reticulum Protein, Is Secreted into the Gastrointestinal Mucus Joakim H. Bergstro¨ m1, Katarina A. Berg1, Ana M. Rodrı´guez-Pin˜ eiro1,Ba¨rbel Stecher2, Malin E. V. Johansson1, Gunnar C. Hansson1* 1 Department of Medical Biochemistry, University of Gothenburg, Gothenburg, Sweden, 2 Max von Pettenkofer Institute for Hygiene and Medical Microbiology, LMU Munich, Munich, Germany Abstract The MUC2 mucin is the major constituent of the two mucus layers in colon. Mice lacking the disulfide isomerase-like protein Agr2 have been shown to be more susceptible to colon inflammation. The Agr22/2 mice have less filled goblet cells and were now shown to have a poorly developed inner colon mucus layer. We could not show AGR2 covalently bound to recombinant MUC2 N- and C-termini as have previously been suggested. We found relatively high concentrations of Agr2 in secreted mucus throughout the murine gastrointestinal tract, suggesting that Agr2 may play extracellular roles. In tissue culture (CHO-K1) cells, AGR2 is normally not secreted. Replacement of the single Cys in AGR2 with Ser (C81S) allowed secretion, suggesting that modification of this Cys might provide a mechanism for circumventing the KTEL endoplasmic reticulum retention signal. In conclusion, these results suggest that AGR2 has both intracellular and extracellular effects in the intestine. Citation: Bergstro¨m JH, Berg KA, Rodrı´guez-Pin˜eiro AM, Stecher B, Johansson MEV, et al. (2014) AGR2, an Endoplasmic Reticulum Protein, Is Secreted into the Gastrointestinal Mucus. PLoS ONE 9(8): e104186. doi:10.1371/journal.pone.0104186 Editor: Jean-Luc Desseyn, Inserm, France Received March 16, 2014; Accepted July 11, 2014; Published August 11, 2014 This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. -

Uterine Double-Conditional Inactivation of Smad2 and Smad3 in Mice Causes Endometrial Dysregulation, Infertility, and Uterine Cancer
Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer Maya Krisemana,b, Diana Monsivaisa,c, Julio Agnoa, Ramya P. Masanda, Chad J. Creightond,e, and Martin M. Matzuka,c,f,g,h,1 aDepartment of Pathology and Immunology, Baylor College of Medicine, Houston, TX 77030; bReproductive Endocrinology and Infertility, Baylor College of Medicine/Texas Children’s Hospital Women’s Pavilion, Houston, TX 77030; cCenter for Drug Discovery, Baylor College of Medicine, Houston, TX 77030; dDepartment of Medicine, Baylor College of Medicine, Houston, TX 77030; eDan L. Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030; fDepartment of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX 77030; gDepartment of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030; and hDepartment of Pharmacology and Chemical Biology, Baylor College of Medicine, Houston, TX 77030 Contributed by Martin M. Matzuk, December 6, 2018 (sent for review April 30, 2018; reviewed by Milan K. Bagchi and Thomas E. Spencer) SMAD2 and SMAD3 are downstream proteins in the transforming in endometrial function. Notably, members of the transforming growth factor-β (TGF β) signaling pathway that translocate signals growth factor β (TGF β) family are involved in many cellular from the cell membrane to the nucleus, bind DNA, and control the processes and serve as principal regulators of numerous biological expression of target genes. While SMAD2/3 have important roles functions, including female reproduction. Previous studies have in the ovary, we do not fully understand the roles of SMAD2/3 in shown the TGF β family to have key roles in ovarian folliculo- the uterus and their implications in the reproductive system. -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -
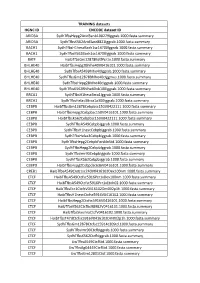
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Differential Physiological Role of BIN1 Isoforms in Skeletal Muscle Development, Function and Regeneration
bioRxiv preprint doi: https://doi.org/10.1101/477950; this version posted December 11, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Differential physiological role of BIN1 isoforms in skeletal muscle development, function and regeneration Ivana Prokic1,2,3,4, Belinda Cowling1,2,3,4, Candice Kutchukian5, Christine Kretz1,2,3,4, Hichem Tasfaout1,2,3,4, Josiane Hergueux1,2,3,4, Olivia Wendling1,2,3,4, Arnaud Ferry10, Anne Toussaint1,2,3,4, Christos Gavriilidis1,2,3,4, Vasugi Nattarayan1,2,3,4, Catherine Koch1,2,3,4, Jeanne Lainné6,7, Roy Combe2,3,4,8, Laurent Tiret9, Vincent Jacquemond5, Fanny Pilot-Storck9, Jocelyn Laporte1,2,3,4 1Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France 2Centre National de la Recherche Scientifique (CNRS), UMR7104, Illkirch, France 3Institut National de la Santé et de la Recherche Médicale (INSERM), U1258, Illkirch, France 4Université de Strasbourg, Illkirch, France 5Univ Lyon, Université Claude Bernard Lyon 1, CNRS UMR-5310, INSERM U-1217, Institut NeuroMyoGène, 8 avenue Rockefeller, 69373 Lyon, France 6Sorbonne Université, INSERM, Institute of Myology, Centre of Research in Myology, UMRS 974, F- 75013, Paris, France 7Sorbonne Université, Department of Physiology, UPMC Univ Paris 06, Pitié-Salpêtrière Hospital, F- 75013, Paris, France 8CELPHEDIA-PHENOMIN, Institut Clinique de la Souris (ICS), Illkirch, France 9U955 – IMRB, Team 10 - Biology of the neuromuscular system, Inserm, UPEC, Ecole nationale vétérinaire d’Alfort, Maisons-Alfort, 94700, France 10Sorbonne Université, INSERM, Institute of Myology, Centre of Research in Myology, UMRS 794, F- 75013, Paris, France Correspondence to: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/477950; this version posted December 11, 2018. -

Cyclin D1 Is a Direct Transcriptional Target of GATA3 in Neuroblastoma Tumor Cells
Oncogene (2010) 29, 2739–2745 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00 www.nature.com/onc SHORT COMMUNICATION Cyclin D1 is a direct transcriptional target of GATA3 in neuroblastoma tumor cells JJ Molenaar1,2, ME Ebus1, J Koster1, E Santo1, D Geerts1, R Versteeg1 and HN Caron2 1Department of Human Genetics, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands and 2Department of Pediatric Oncology, Emma Kinderziekenhuis, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands Almost all neuroblastoma tumors express excess levels of 2000). Several checkpoints normally prevent premature Cyclin D1 (CCND1) compared to normal tissues and cell-cycle progression and cell division. The crucial G1 other tumor types. Only a small percentage of these entry point is controlled by the D-type Cyclins that can neuroblastoma tumors have high-level amplification of the activate CDK4/6 that in turn phosphorylate the pRb Cyclin D1 gene. The other neuroblastoma tumors have protein. This results in a release of the E2F transcription equally high Cyclin D1 expression without amplification. factor that causes transcriptional upregulation of Silencing of Cyclin D1 expression was previously found to numerous genes involved in further progression of the trigger differentiation of neuroblastoma cells. Over- cell cycle (Sherr, 1996). expression of Cyclin D1 is therefore one of the most Neuroblastomas are embryonal tumors that originate frequent mechanisms with a postulated function in neuro- from precursor cells of the sympathetic nervous system. blastoma pathogenesis. The cause for the Cyclin D1 This tumor has a very poor prognosis and despite the overexpression is unknown. -

SUPPLEMENTAL DATA Supplemental Materials And
SUPPLEMENTAL DATA Supplemental Materials and Methods Cells and Cell Culture Human breast carcinoma cell lines, MDA-MB-231 and MCF7, were purchased from American Type Tissue Culture Collection (ATCC). 231BoM-1833, 231BrM-2a, CN34, CN34-BoM2d, CN34-BrM2c and MCF7- BoM2d cell lines were kindly provided by Dr. Joan Massagué (Memorial Sloan-Kettering Cancer Center) (1-3). Luciferase-labeled cells were generated by infecting the lentivirus carrying the firefly luciferase gene. The immortalized mouse bone microvascular endothelial cell (mBMEC) was a generous gift from Dr. Isaiah J. Fidler (M.D. Anderson Cancer Center) (4). MCF10A and MCF10DCIS.com cells were purchased from ATCC and Asterand, respectively. MDA-MB-231, its variant cells, MCF7 and MCF-BoM2d cells were cultured in DMEM medium supplemented with 10% FBS and antibiotics. CN34 and its variant cells were cultured in Medium199 supplemented with 2.5% FBS, 10 µg/ml insulin, 0.5 µg/ml hydrocortisone, 20 ng/ml EGF, 100 ng/ml cholera toxin and antibiotics. MCF10DCIS.com cells were cultured in RPMI-1640 medium supplemented with 10% FBS and antibiotics. MCF10A cells were cultured in MEGM mammary epithelial cell growth medium (Lonza). mBMEC was maintained at 8% CO2 at 33 °C in DMEM with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids and 1% vitamin mixture. Bone marrow stromal fibroblast cell lines HS5 and HS27A, and osteoblast cell line, hFOB1.19, were purchased from ATCC. Bone marrow derived human mesenchymal stem cells, BM-hMSC, were isolated for enrichment of plastic adherent cells from unprocessed bone marrow (Lonza) which was depleted of red blood cells. -

Supplementary Table S1. Upregulated Genes Differentially
Supplementary Table S1. Upregulated genes differentially expressed in athletes (p < 0.05 and 1.3-fold change) Gene Symbol p Value Fold Change 221051_s_at NMRK2 0.01 2.38 236518_at CCDC183 0.00 2.05 218804_at ANO1 0.00 2.05 234675_x_at 0.01 2.02 207076_s_at ASS1 0.00 1.85 209135_at ASPH 0.02 1.81 228434_at BTNL9 0.03 1.81 229985_at BTNL9 0.01 1.79 215795_at MYH7B 0.01 1.78 217979_at TSPAN13 0.01 1.77 230992_at BTNL9 0.01 1.75 226884_at LRRN1 0.03 1.74 220039_s_at CDKAL1 0.01 1.73 236520_at 0.02 1.72 219895_at TMEM255A 0.04 1.72 201030_x_at LDHB 0.00 1.69 233824_at 0.00 1.69 232257_s_at 0.05 1.67 236359_at SCN4B 0.04 1.64 242868_at 0.00 1.63 1557286_at 0.01 1.63 202780_at OXCT1 0.01 1.63 1556542_a_at 0.04 1.63 209992_at PFKFB2 0.04 1.63 205247_at NOTCH4 0.01 1.62 1554182_at TRIM73///TRIM74 0.00 1.61 232892_at MIR1-1HG 0.02 1.61 204726_at CDH13 0.01 1.6 1561167_at 0.01 1.6 1565821_at 0.01 1.6 210169_at SEC14L5 0.01 1.6 236963_at 0.02 1.6 1552880_at SEC16B 0.02 1.6 235228_at CCDC85A 0.02 1.6 1568623_a_at SLC35E4 0.00 1.59 204844_at ENPEP 0.00 1.59 1552256_a_at SCARB1 0.02 1.59 1557283_a_at ZNF519 0.02 1.59 1557293_at LINC00969 0.03 1.59 231644_at 0.01 1.58 228115_at GAREM1 0.01 1.58 223687_s_at LY6K 0.02 1.58 231779_at IRAK2 0.03 1.58 243332_at LOC105379610 0.04 1.58 232118_at 0.01 1.57 203423_at RBP1 0.02 1.57 AMY1A///AMY1B///AMY1C///AMY2A///AMY2B// 208498_s_at 0.03 1.57 /AMYP1 237154_at LOC101930114 0.00 1.56 1559691_at 0.01 1.56 243481_at RHOJ 0.03 1.56 238834_at MYLK3 0.01 1.55 213438_at NFASC 0.02 1.55 242290_at TACC1 0.04 1.55 ANKRD20A1///ANKRD20A12P///ANKRD20A2/// -

Study of Natural Longlife Juvenility and Tissue Regeneration in Caudate Amphibians and Potential Application of Resulting Data in Biomedicine
Journal of Developmental Biology Review Study of Natural Longlife Juvenility and Tissue Regeneration in Caudate Amphibians and Potential Application of Resulting Data in Biomedicine Eleonora N. Grigoryan Kol’tsov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia; [email protected]; Tel.: +7-(499)-1350052 Abstract: The review considers the molecular, cellular, organismal, and ontogenetic properties of Urodela that exhibit the highest regenerative abilities among tetrapods. The genome specifics and the expression of genes associated with cell plasticity are analyzed. The simplification of tissue structure is shown using the examples of the sensory retina and brain in mature Urodela. Cells of these and some other tissues are ready to initiate proliferation and manifest the plasticity of their phenotype as well as the correct integration into the pre-existing or de novo forming tissue structure. Without excluding other factors that determine regeneration, the pedomorphosis and juvenile properties, identified on different levels of Urodele amphibians, are assumed to be the main explanation for their high regenerative abilities. These properties, being fundamental for tissue regeneration, have been lost by amniotes. Experiments aimed at mammalian cell rejuvenation currently use various approaches. They include, in particular, methods that use secretomes from regenerating tissues of caudate amphibians and fish for inducing regenerative responses of cells. Such an approach, along with those developed on the basis of knowledge about the molecular and genetic nature and age dependence of regeneration, may become one more step in the development of regenerative medicine Citation: Grigoryan, E.N. Study of Keywords: salamanders; juvenile state; tissue regeneration; extracts; microvesicles; cell rejuvenation Natural Longlife Juvenility and Tissue Regeneration in Caudate Amphibians and Potential Application of Resulting Data in 1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Clinicopathological and Molecular Genetic Analysis of Low-Grade Glioma in Adults
A CLINICOPATHOLOGICAL AND MOLECULAR GENETIC ANALYSIS OF LOW-GRADE GLIOMA IN ADULTS Presented by ANUSHREE SINGH MSc A thesis submitted in partial fulfilment of the requirements of the University of Wolverhampton for the degree of Doctor of Philosophy Brain Tumour Research Centre Research Institute in Healthcare Sciences Faculty of Science and Engineering University of Wolverhampton November 2014 i DECLARATION This work or any part thereof has not previously been presented in any form to the University or to any other body whether for the purposes of assessment, publication or for any other purpose (unless otherwise indicated). Save for any express acknowledgments, references and/or bibliographies cited in the work, I confirm that the intellectual content of the work is the result of my own efforts and of no other person. The right of Anushree Singh to be identified as author of this work is asserted in accordance with ss.77 and 78 of the Copyright, Designs and Patents Act 1988. At this date copyright is owned by the author. Signature: Anushree Date: 30th November 2014 ii ABSTRACT The aim of the study was to identify molecular markers that can determine progression of low grade glioma. This was done using various approaches such as IDH1 and IDH2 mutation analysis, MGMT methylation analysis, copy number analysis using array comparative genomic hybridisation and identification of differentially expressed miRNAs using miRNA microarray analysis. IDH1 mutation was present at a frequency of 71% in low grade glioma and was identified as an independent marker for improved OS in a multivariate analysis, which confirms the previous findings in low grade glioma studies. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between