The Alzheimer's Disease Genetic Risk Factor BIN1 Induces Isoform
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Differential Physiological Role of BIN1 Isoforms in Skeletal Muscle Development, Function and Regeneration
bioRxiv preprint doi: https://doi.org/10.1101/477950; this version posted December 11, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Differential physiological role of BIN1 isoforms in skeletal muscle development, function and regeneration Ivana Prokic1,2,3,4, Belinda Cowling1,2,3,4, Candice Kutchukian5, Christine Kretz1,2,3,4, Hichem Tasfaout1,2,3,4, Josiane Hergueux1,2,3,4, Olivia Wendling1,2,3,4, Arnaud Ferry10, Anne Toussaint1,2,3,4, Christos Gavriilidis1,2,3,4, Vasugi Nattarayan1,2,3,4, Catherine Koch1,2,3,4, Jeanne Lainné6,7, Roy Combe2,3,4,8, Laurent Tiret9, Vincent Jacquemond5, Fanny Pilot-Storck9, Jocelyn Laporte1,2,3,4 1Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Illkirch, France 2Centre National de la Recherche Scientifique (CNRS), UMR7104, Illkirch, France 3Institut National de la Santé et de la Recherche Médicale (INSERM), U1258, Illkirch, France 4Université de Strasbourg, Illkirch, France 5Univ Lyon, Université Claude Bernard Lyon 1, CNRS UMR-5310, INSERM U-1217, Institut NeuroMyoGène, 8 avenue Rockefeller, 69373 Lyon, France 6Sorbonne Université, INSERM, Institute of Myology, Centre of Research in Myology, UMRS 974, F- 75013, Paris, France 7Sorbonne Université, Department of Physiology, UPMC Univ Paris 06, Pitié-Salpêtrière Hospital, F- 75013, Paris, France 8CELPHEDIA-PHENOMIN, Institut Clinique de la Souris (ICS), Illkirch, France 9U955 – IMRB, Team 10 - Biology of the neuromuscular system, Inserm, UPEC, Ecole nationale vétérinaire d’Alfort, Maisons-Alfort, 94700, France 10Sorbonne Université, INSERM, Institute of Myology, Centre of Research in Myology, UMRS 794, F- 75013, Paris, France Correspondence to: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/477950; this version posted December 11, 2018. -

Murine Megakaryopoiesis Is Critical for P21 SCL-Mediated Regulation Of
From bloodjournal.hematologylibrary.org at UNIVERSITY OF BIRMINGHAM on March 1, 2012. For personal use only. 2011 118: 723-735 Prepublished online May 19, 2011; doi:10.1182/blood-2011-01-328765 SCL-mediated regulation of the cell-cycle regulator p21 is critical for murine megakaryopoiesis Hedia Chagraoui, Mira Kassouf, Sreemoti Banerjee, Nicolas Goardon, Kevin Clark, Ann Atzberger, Andrew C. Pearce, Radek C. Skoda, David J. P. Ferguson, Steve P. Watson, Paresh Vyas and Catherine Porcher Updated information and services can be found at: http://bloodjournal.hematologylibrary.org/content/118/3/723.full.html Articles on similar topics can be found in the following Blood collections Platelets and Thrombopoiesis (260 articles) Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved. From bloodjournal.hematologylibrary.org at UNIVERSITY OF BIRMINGHAM on March 1, 2012. For personal use only. PLATELETS AND THROMBOPOIESIS SCL-mediated regulation of the cell-cycle regulator p21 is critical for murine megakaryopoiesis Hedia Chagraoui,1 *Mira Kassouf,1 *Sreemoti Banerjee,1 Nicolas Goardon,1 Kevin Clark,1 Ann Atzberger,1 Andrew C. -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Supplementary Table 8. Cpcp PPI Network Details for Significantly Changed Proteins, As Identified in 3.2, Underlying Each of the Five Functional Domains
Supplementary Table 8. cPCP PPI network details for significantly changed proteins, as identified in 3.2, underlying each of the five functional domains. The network nodes represent each significant protein, followed by the list of interactors. Note that identifiers were converted to gene names to facilitate PPI database queries. Functional Domain Node Interactors Development and Park7 Rack1 differentiation Kcnma1 Atp6v1a Ywhae Ywhaz Pgls Hsd3b7 Development and Prdx6 Ncoa3 differentiation Pla2g4a Sufu Ncf2 Gstp1 Grin2b Ywhae Pgls Hsd3b7 Development and Atp1a2 Kcnma1 differentiation Vamp2 Development and Cntn1 Prnp differentiation Ywhaz Clstn1 Dlg4 App Ywhae Ywhab Development and Rac1 Pak1 differentiation Cdc42 Rhoa Dlg4 Ctnnb1 Mapk9 Mapk8 Pik3cb Sod1 Rrad Epb41l2 Nono Ltbp1 Evi5 Rbm39 Aplp2 Smurf2 Grin1 Grin2b Xiap Chn2 Cav1 Cybb Pgls Ywhae Development and Hbb-b1 Atp5b differentiation Hba Kcnma1 Got1 Aldoa Ywhaz Pgls Hsd3b4 Hsd3b7 Ywhae Development and Myh6 Mybpc3 differentiation Prkce Ywhae Development and Amph Capn2 differentiation Ap2a2 Dnm1 Dnm3 Dnm2 Atp6v1a Ywhab Development and Dnm3 Bin1 differentiation Amph Pacsin1 Grb2 Ywhae Bsn Development and Eef2 Ywhaz differentiation Rpgrip1l Atp6v1a Nphp1 Iqcb1 Ezh2 Ywhae Ywhab Pgls Hsd3b7 Hsd3b4 Development and Gnai1 Dlg4 differentiation Development and Gnao1 Dlg4 differentiation Vamp2 App Ywhae Ywhab Development and Psmd3 Rpgrip1l differentiation Psmd4 Hmga2 Development and Thy1 Syp differentiation Atp6v1a App Ywhae Ywhaz Ywhab Hsd3b7 Hsd3b4 Development and Tubb2a Ywhaz differentiation Nphp4 -

BIN1 Gene Bridging Integrator 1
BIN1 gene bridging integrator 1 Normal Function The BIN1 gene provides instructions for making a protein that is found in tissues throughout the body, where it interacts with a variety of other proteins. The BIN1 protein is thought to be involved in the transportation of materials from the cell surface into the cell (endocytosis) and the self-destruction of cells (apoptosis). The BIN1 protein may act as a tumor suppressor protein, which means it prevents cells from growing and dividing too rapidly or in an uncontrolled way. Several different versions (isoforms) of the BIN1 protein are produced from the BIN1 gene. These isoforms vary by size and are active in different tissues. The BIN1 protein isoform that is expressed in muscle cells is thought to be involved in the formation of structures called transverse tubules or T tubules. These structures are found within the membrane of muscle cells, where they play a role in muscle tensing (contraction) and relaxation. Health Conditions Related to Genetic Changes Centronuclear myopathy At least 10 mutations in the BIN1 gene have been found to cause centronuclear myopathy, a condition that is characterized by muscle weakness (myopathy) in the skeletal muscles, which are the muscles used for movement. Most of these mutations change single protein building blocks (amino acids) in the BIN1 protein. BIN1 gene mutations result in the production of a protein that cannot form T tubules. A shortage of T tubules in muscle fibers alters their structure, which prevents them from contracting and relaxing normally. The abnormal muscle fibers underlie the muscle weakness characteristic of centronuclear myopathy. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

WO 2017/004142 Al Oo
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/004142 Al 5 January 2017 (05.01.2017) P O P C T (51) International Patent Classification: (71) Applicants: CHILDREN'S MEDICAL CENTER COR¬ A61P 21/00 (2006.01) C12N 15/113 (2010.01) PORATION [US/US]; 55 Shattuck Street, Boston, MA A61K 39/395 (2006.01) C07K 16/18 (2006.01) 021 15 (US). PRESIDENT AND FELLOWS OF HAR¬ A61K 31/713 (2006.01) VARD COLLEGE [US/US]; 17 Quincy Street, Cam bridge, MA 021 38 (US). (21) International Application Number: PCT/US20 16/039998 (72) Inventors: SMITH, Laura, L.; 523 Columbus Ave Apt. G, Boston, MA 021 18 (US). GUPTA, Vandana; 2 Bates (22) International Filing Date: Road, Lexington, MA 02421 (US). BEGGS, Alan; 120 29 June 2016 (29.06.2016) High Rock Street, Needham, MA 02492 (US). (25) Filing Language: English (74) Agent: EL-HAYEK, Roque; Wolf, Greenfield & Sacks, (26) Publication Language: English P.C., 600 Atlantic Avenue, Boston, MA 02210-2206 (US). (30) Priority Data: (81) Designated States (unless otherwise indicated, for every 62/185,760 29 June 2015 (29.06.2015) US kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, [Continued on nextpage] (54) Title: TREATMENT FOR MYOPATHY Abstract: Provided herein are methods and compositions for the treatment of myopathies. ryrlb 'i , ryri FIG. -

Alzheimer's Disease Risk Gene BIN1 Induces Tau-Dependent Network
RESEARCH ARTICLE Alzheimer’s disease risk gene BIN1 induces Tau-dependent network hyperexcitability Yuliya Voskobiynyk1†, Jonathan R Roth1†, J Nicholas Cochran1, Travis Rush1, Nancy VN Carullo2, Jacob S Mesina1, Mohammad Waqas1, Rachael M Vollmer1, Jeremy J Day2, Lori L McMahon3, Erik D Roberson1* 1Center for Neurodegeneration and Experimental Therapeutics, Alzheimer’s Disease Center, and Evelyn F. McKnight Brain Institute, Departments of Neurology and Neurobiology, University of Alabama at Birmingham, Birmingham, United States; 2Department of Neurobiology, University of Alabama at Birmingham, Birmingham, United States; 3Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, United States Abstract Genome-wide association studies identified the BIN1 locus as a leading modulator of genetic risk in Alzheimer’s disease (AD). One limitation in understanding BIN1’s contribution to AD is its unknown function in the brain. AD-associated BIN1 variants are generally noncoding and likely change expression. Here, we determined the effects of increasing expression of the major neuronal isoform of human BIN1 in cultured rat hippocampal neurons. Higher BIN1 induced network hyperexcitability on multielectrode arrays, increased frequency of synaptic transmission, and elevated calcium transients, indicating that increasing BIN1 drives greater neuronal activity. In exploring the mechanism of these effects on neuronal physiology, we found that BIN1 interacted with L-type voltage-gated calcium channels (LVGCCs) and that BIN1–LVGCC interactions were modulated by Tau in rat hippocampal neurons and mouse brain. Finally, Tau reduction prevented BIN1-induced network hyperexcitability. These data shed light on BIN1’s neuronal function and *For correspondence: suggest that it may contribute to Tau-dependent hyperexcitability in AD. -

Supplementary Files:To “Allelic Switching of DLX5, GRB10 and SVOPL During Colorectal
Supplementary files:to “Allelic switching of DLX5, GRB10 and SVOPL during colorectal cancer tumorigenesis” Arnoud Boot, Jan Oosting, Saskia Doorn, Sarah Ouahoud, Marina Ventayol Garcia, Dina Ruano, Hans Morreau, Tom van Wezel* Supplementary figure 1: ASE scores for selected chromosome 7 genes as determined by cSNP arrays. ASE scores were only calculated for cell lines with a heterozygous cSNP in the DNA. Supplementary figure 2: Gene expression of SVOPL and DLX5. Supplementary table 1: ASE results per cell line for all genes with at least 1 heterozygous sample Per sample, each gene was assigned an ASE score, based on the data from the heterozygous cSNPs, as described in the Supplementary methods. Samples displaying ASE for that gene are marked 1, samples not showing ASE are marked 0. For samples where no heterozygous cSNPs were identified, the field was left empty. Genes for which none of the samples showed a heterozygous cSNP were removed, as ASE could not be calculated. Supplementary table 2: DLX5 ASE results in paired normal and cancer samples. ASE scores for paired normal mucosa and colorectal cancer samples. The 4th column states the case- level conclusion concerning allelic switching during tumorigenesis. Supplementary table 3: PRPS1L1 KASPar genotyping results in paired normal and cancer samples. Supplementary table 4: Samples used in this study. Supplementary methods: ASE detection method and array analysis QC Figure S1: ASE scores for selected chromosome 7 genes Figure S2: Gene expression of SVOPL and DLX5 Supplementary methods: ASE detection method and array analysis QC Detection of ASE was performed by hybridizing both DNA and cDNA to the Infinium HumanExome- 12v1 beadchips. -

The Kinesin Spindle Protein Inhibitor Filanesib Enhances the Activity of Pomalidomide and Dexamethasone in Multiple Myeloma
Plasma Cell Disorders SUPPLEMENTARY APPENDIX The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma Susana Hernández-García, 1 Laura San-Segundo, 1 Lorena González-Méndez, 1 Luis A. Corchete, 1 Irena Misiewicz- Krzeminska, 1,2 Montserrat Martín-Sánchez, 1 Ana-Alicia López-Iglesias, 1 Esperanza Macarena Algarín, 1 Pedro Mogollón, 1 Andrea Díaz-Tejedor, 1 Teresa Paíno, 1 Brian Tunquist, 3 María-Victoria Mateos, 1 Norma C Gutiérrez, 1 Elena Díaz- Rodriguez, 1 Mercedes Garayoa 1* and Enrique M Ocio 1* 1Centro Investigación del Cáncer-IBMCC (CSIC-USAL) and Hospital Universitario-IBSAL, Salamanca, Spain; 2National Medicines Insti - tute, Warsaw, Poland and 3Array BioPharma, Boulder, Colorado, USA *MG and EMO contributed equally to this work ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.168666 Received: March 13, 2017. Accepted: August 29, 2017. Pre-published: August 31, 2017. Correspondence: [email protected] MATERIAL AND METHODS Reagents and drugs. Filanesib (F) was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide (T), lenalidomide (L) and pomalidomide (P) were purchased from Selleckchem (Houston, TX, USA), dexamethasone (D) from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA). Generic chemicals were acquired from Sigma Chemical Co., Roche Biochemicals (Mannheim, Germany), Merck & Co., Inc. (Darmstadt, Germany). MM cell lines, patient samples and cultures. Origin, authentication and in vitro growth conditions of human MM cell lines have already been characterized (17, 18). The study of drug activity in the presence of IL-6, IGF-1 or in co-culture with primary bone marrow mesenchymal stromal cells (BMSCs) or the human mesenchymal stromal cell line (hMSC–TERT) was performed as described previously (19, 20). -

Roles of Splicing Factors in Hormone-Related Cancer Progression
International Journal of Molecular Sciences Review Roles of Splicing Factors in Hormone-Related Cancer Progression Toshihiko Takeiwa 1, Yuichi Mitobe 1, Kazuhiro Ikeda 1, Kuniko Horie-Inoue 1 and Satoshi Inoue 1,2,* 1 Division of Gene Regulation and Signal Transduction, Research Center for Genomic Medicine, Saitama Medical University, Hidaka, Saitama 350-1241, Japan; [email protected] (T.T.); [email protected] (Y.M.); [email protected] (K.I.); [email protected] (K.H.-I.) 2 Department of Systems Aging Science and Medicine, Tokyo Metropolitan Institute of Gerontology, Itabashi-ku, Tokyo 173-0015, Japan * Correspondence: [email protected]; Tel.: +81-3-3964-3241 Received: 8 February 2020; Accepted: 20 February 2020; Published: 25 February 2020 Abstract: Splicing of mRNA precursor (pre-mRNA) is a mechanism to generate multiple mRNA isoforms from a single pre-mRNA, and it plays an essential role in a variety of biological phenomena and diseases such as cancers. Previous studies have demonstrated that cancer-specific splicing events are involved in various aspects of cancers such as proliferation, migration and response to hormones, suggesting that splicing-targeting therapy can be promising as a new strategy for cancer treatment. In this review, we focus on the splicing regulation by RNA-binding proteins including Drosophila behavior/human splicing (DBHS) family proteins, serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) in hormone-related cancers, such as breast and prostate cancers. Keywords: DBHS family proteins; SR proteins; hnRNPs; breast cancer; prostate cancer 1. Introduction Splicing of mRNA precursors (pre-mRNAs) is an essential mechanism in the posttranscriptional regulation of gene expression. -
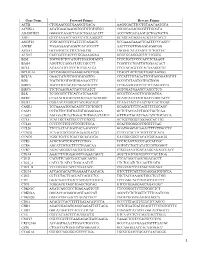
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC