1 Title: Genome-Wide Map of Regulatory Interactions in the Human
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table S1. List of Proteins in the BAHD1 Interactome
Table S1. List of proteins in the BAHD1 interactome BAHD1 nuclear partners found in this work yeast two-hybrid screen Name Description Function Reference (a) Chromatin adapters HP1α (CBX5) chromobox homolog 5 (HP1 alpha) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins (20-23) HP1β (CBX1) chromobox homolog 1 (HP1 beta) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins HP1γ (CBX3) chromobox homolog 3 (HP1 gamma) Binds histone H3 methylated on lysine 9 and chromatin-associated proteins MBD1 methyl-CpG binding domain protein 1 Binds methylated CpG dinucleotide and chromatin-associated proteins (22, 24-26) Chromatin modification enzymes CHD1 chromodomain helicase DNA binding protein 1 ATP-dependent chromatin remodeling activity (27-28) HDAC5 histone deacetylase 5 Histone deacetylase activity (23,29,30) SETDB1 (ESET;KMT1E) SET domain, bifurcated 1 Histone-lysine N-methyltransferase activity (31-34) Transcription factors GTF3C2 general transcription factor IIIC, polypeptide 2, beta 110kDa Required for RNA polymerase III-mediated transcription HEYL (Hey3) hairy/enhancer-of-split related with YRPW motif-like DNA-binding transcription factor with basic helix-loop-helix domain (35) KLF10 (TIEG1) Kruppel-like factor 10 DNA-binding transcription factor with C2H2 zinc finger domain (36) NR2F1 (COUP-TFI) nuclear receptor subfamily 2, group F, member 1 DNA-binding transcription factor with C4 type zinc finger domain (ligand-regulated) (36) PEG3 paternally expressed 3 DNA-binding transcription factor with -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -
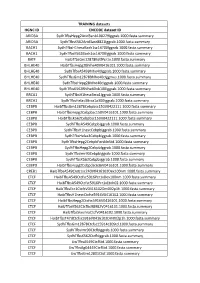
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Prospective Isolation of NKX2-1–Expressing Human Lung Progenitors Derived from Pluripotent Stem Cells
The Journal of Clinical Investigation RESEARCH ARTICLE Prospective isolation of NKX2-1–expressing human lung progenitors derived from pluripotent stem cells Finn Hawkins,1,2 Philipp Kramer,3 Anjali Jacob,1,2 Ian Driver,4 Dylan C. Thomas,1 Katherine B. McCauley,1,2 Nicholas Skvir,1 Ana M. Crane,3 Anita A. Kurmann,1,5 Anthony N. Hollenberg,5 Sinead Nguyen,1 Brandon G. Wong,6 Ahmad S. Khalil,6,7 Sarah X.L. Huang,3,8 Susan Guttentag,9 Jason R. Rock,4 John M. Shannon,10 Brian R. Davis,3 and Darrell N. Kotton1,2 2 1Center for Regenerative Medicine, and The Pulmonary Center and Department of Medicine, Boston University School of Medicine, Boston, Massachusetts, USA. 3Center for Stem Cell and Regenerative Medicine, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center, Houston, Texas, USA. 4Department of Anatomy, UCSF, San Francisco, California, USA. 5Division of Endocrinology, Diabetes and Metabolism, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts, USA. 6Department of Biomedical Engineering and Biological Design Center, Boston University, Boston, Massachusetts, USA. 7Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts, USA. 8Columbia Center for Translational Immunology & Columbia Center for Human Development, Columbia University Medical Center, New York, New York, USA. 9Department of Pediatrics, Monroe Carell Jr. Children’s Hospital, Vanderbilt University, Nashville, Tennessee, USA. 10Division of Pulmonary Biology, Cincinnati Children’s Hospital, Cincinnati, Ohio, USA. It has been postulated that during human fetal development, all cells of the lung epithelium derive from embryonic, endodermal, NK2 homeobox 1–expressing (NKX2-1+) precursor cells. -

Feedback Regulation Between Initiation and Maturation Networks Orchestrates the Chromatin Dynamics of Epidermal Lineage
bioRxiv preprint doi: https://doi.org/10.1101/349308; this version posted June 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Li et al., p. 1 Feedback Regulation between Initiation and Maturation Networks Orchestrates the Chromatin Dynamics of Epidermal Lineage Commitment Lingjie Li1,3,4, Yong Wang2,4,7,8*, Jessica L. Torkelson1,3*, Gautam Shankar1, Jillian M. Pattison1,3, Hanson H. Zhen1,3, Zhana Duren2,4,7, Fengqin Fang5, Sandra P. Melo1, Samantha N. Piekos1,3, Jiang Li1, Eric J. Liaw1, Lang Chen7, Rui Li1,4, Marius Wernig6, Wing H. Wong2,4, Howard Y. Chang1,4, Anthony E. Oro1,3,9 1 Program in Epithelial Biology and Department of Dermatology 2 Department of Statistics and Biomedical Data Science 3 Center for Definitive and Curative Medicine 4 Center for Personal Dynamic Regulome 5 Division of Immunology and Rheumatology, Department of Medicine, 6 Institute for Stem Cell Biology and Regenerative Medicine, Department of Pathology, Stanford University School of Medicine, Stanford, CA 94305, USA. 7 CEMS, NCMIS, MDIS, Academy of Mathematics & Systems Science, Chinese Academy of Sciences, Beijing,100080, China 8 Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, 650223, China *These authors made equal and independent contributions. 9 Correspondence to Lead Contact: Anthony E. Oro at [email protected] bioRxiv preprint doi: https://doi.org/10.1101/349308; this version posted June 18, 2018. -

The Activator Protein-1 Transcription Factor in Respiratory Epithelium Carcinogenesis
Subject Review The Activator Protein-1 Transcription Factor in Respiratory Epithelium Carcinogenesis Michalis V. Karamouzis,1 Panagiotis A. Konstantinopoulos,1,2 and Athanasios G. Papavassiliou1 1Department of Biological Chemistry, Medical School, University of Athens, Athens, Greece and 2Division of Hematology-Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts Abstract Much of the current anticancer research effort is focused on Respiratory epithelium cancers are the leading cause cell-surface receptors and their cognate upstream molecules of cancer-related death worldwide. The multistep natural because they provide the easiest route for drugs to affect history of carcinogenesis can be considered as a cellular behavior, whereas agents acting at the level of gradual accumulation of genetic and epigenetic transcription need to invade the nucleus. However, the aberrations, resulting in the deregulation of cellular therapeutic effect of surface receptor manipulation might be homeostasis. Growing evidence suggests that cross- considered less than specific because their actions are talk between membrane and nuclear receptor signaling modulated by complex interacting downstream signal trans- pathways along with the activator protein-1 (AP-1) duction pathways. A pivotal transcription factor during cascade and its cofactor network represent a pivotal respiratory epithelium carcinogenesis is activator protein-1 molecular circuitry participating directly or indirectly in (AP-1). AP-1–regulated genes include important modulators of respiratory epithelium carcinogenesis. The crucial role invasion and metastasis, proliferation, differentiation, and of AP-1 transcription factor renders it an appealing survival as well as genes associated with hypoxia and target of future nuclear-directed anticancer therapeutic angiogenesis (7). Nuclear-directed therapeutic strategies might and chemoprevention approaches. -

To Study Mutant P53 Gain of Function, Various Tumor-Derived P53 Mutants
Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By Shama K Khokhar M.Sc., Bilaspur University, 2004 B.Sc., Bhopal University, 2002 2007 1 COPYRIGHT SHAMA K KHOKHAR 2007 2 WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES Date of Defense: 12-03-07 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY SHAMA KHAN KHOKHAR ENTITLED Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science Madhavi P. Kadakia, Ph.D. Thesis Director Daniel Organisciak , Ph.D. Department Chair Committee on Final Examination Madhavi P. Kadakia, Ph.D. Steven J. Berberich, Ph.D. Michael Leffak, Ph.D. Joseph F. Thomas, Jr., Ph.D. Dean, School of Graduate Studies 3 Abstract Khokhar, Shama K. M.S., Department of Biochemistry and Molecular Biology, Wright State University, 2007 Differential effect of TAp63γ mutants on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. p63, a member of the p53 gene family, known to play a role in development, has more recently also been implicated in cancer progression. Mice lacking p63 exhibit severe developmental defects such as limb truncations, abnormal skin, and absence of hair follicles, teeth, and mammary glands. Germline missense mutations of p63 have been shown to be responsible for several human developmental syndromes including SHFM, EEC and ADULT syndromes and are associated with anomalies in the development of organs of epithelial origin. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

The Alternative Role of DNA Methylation in Splicing Regulation
TIGS-1191; No. of Pages 7 Review The alternative role of DNA methylation in splicing regulation Galit Lev Maor, Ahuvi Yearim, and Gil Ast Department of Human Molecular Genetics and Biochemistry, Sackler Medical School, Tel Aviv University, Tel Aviv, Israel Although DNA methylation was originally thought to only Alternative splicing is an evolutionarily conserved mech- affect transcription, emerging evidence shows that it also anism that increases transcriptome and proteome diversity regulates alternative splicing. Exons, and especially splice by allowing the generation of multiple mRNA products from sites, have higher levels of DNA methylation than flanking a single gene [10]. More than 90% of human genes were introns, and the splicing of about 22% of alternative exons shown to undergo alternative splicing [11,12]. Furthermore, is regulated by DNA methylation. Two different mecha- the average number of spliced isoforms per gene is higher in nisms convey DNA methylation information into the vertebrates [13], implying that the prevalence of alternative regulation of alternative splicing. The first involves mod- splicing in these organisms is important for their greater ulation of the elongation rate of RNA polymerase II (Pol II) complexity. The splicing reaction is regulated by various by CCCTC-binding factor (CTCF) and methyl-CpG binding activating and repressing elements such as cis-acting se- protein 2 (MeCP2); the second involves the formation of a quence signals and RNA-binding proteins [13–15]. Its regu- protein bridge by heterochromatin protein 1 (HP1) that lation is essential for providing cells and tissues their recruits splicing factors onto transcribed alternative specific features, and for their response to environmental exons. -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution. -

Hepatitis C Virus As a Unique Human Model Disease to Define
viruses Review Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection David Wolski and Georg M. Lauer * Liver Center at the Gastrointestinal Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA * Correspondence: [email protected]; Tel.: +1-617-724-7515 Received: 27 June 2019; Accepted: 23 July 2019; Published: 26 July 2019 Abstract: The hepatitis C virus is unique among chronic viral infections in that an acute outcome with complete viral elimination is observed in a minority of infected patients. This unique feature allows direct comparison of successful immune responses with those that fail in the setting of the same human infection. Here we review how this scenario can be used to achieve better understanding of transcriptional regulation of T-cell differentiation. Specifically, we discuss results from a study comparing transcriptional profiles of hepatitis C virus (HCV)-specific CD8 T-cells during early HCV infection between patients that do and do not control and eliminate HCV. Identification of early gene expression differences in key T-cell differentiation molecules as well as clearly distinct transcriptional networks related to cell metabolism and nucleosomal regulation reveal novel insights into the development of exhausted and memory T-cells. With additional transcriptional studies of HCV-specific CD4 and CD8 T-cells in different stages of infection currently underway, we expect HCV infection to become a valuable model disease to study human immunity to viruses. Keywords: viral hepatitis; hepatitis C virus; T cells; transcriptional regulation; transcription factors; metabolism; nucleosome 1.