Feedback Regulation Between Initiation and Maturation Networks Orchestrates the Chromatin Dynamics of Epidermal Lineage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Unfolded Protein Response: an Overview
biology Review The Unfolded Protein Response: An Overview Adam Read 1,2 and Martin Schröder 1,2,* 1 Department of Biosciences, Durham University, South Road, Durham DH1 3LE, UK; [email protected] 2 Biophysical Sciences Institute, Durham University, South Road, Durham DH1 3LE, UK * Correspondence: [email protected]; Tel.: +44-191-334-1316 Simple Summary: The unfolded protein response (UPR) is the cells’ way of maintaining the balance of protein folding in the endoplasmic reticulum, which is the section of the cell designated for folding proteins with specific destinations such as other organelles or to be secreted by the cell. The UPR is activated when unfolded proteins accumulate in the endoplasmic reticulum. This accumulation puts a greater load on the molecules in charge of folding the proteins, and therefore the UPR works to balance this by lowering the number of unfolded proteins present in the cell. This is done in multiple ways, such as lowering the number of proteins that need to be folded; increasing the folding ability of the endoplasmic reticulum and by removing some of the unfolded proteins which take longer to fold. If the UPR is successful at reducing the number of unfolded proteins, the UPR is inactivated and the cells protein folding balance is returned to normal. However, if the UPR is unsuccessful, then this can lead to cell death. Abstract: The unfolded protein response is the mechanism by which cells control endoplasmic reticulum (ER) protein homeostasis. Under normal conditions, the UPR is not activated; however, under certain stresses, such as hypoxia or altered glycosylation, the UPR can be activated due to an accumulation of unfolded proteins. -

Combinatorial Bzip Dimers Display Complex DNA-Binding Specificity Landscapes
Combinatorial bZIP dimers display complex DNA-binding specificity landscapes The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rodriguez-Martinez, Jose A et al. “Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes.” eLife 6 (2017): n. pag. As Published http://dx.doi.org/10.7554/eLife.19272 Publisher eLife Sciences Publications, Ltd. Version Final published version Citable link http://hdl.handle.net/1721.1/110147 Terms of Use Creative Commons Attribution 4.0 International License Detailed Terms http://creativecommons.org/licenses/by-nc/4.0/ RESEARCH ARTICLE Combinatorial bZIP dimers display complex DNA-binding specificity landscapes Jose´ A Rodrı´guez-Martı´nez1†, Aaron W Reinke2†, Devesh Bhimsaria1,3†, Amy E Keating2,4, Aseem Z Ansari1,5* 1Department of Biochemistry, University of Wisconsin-Madison, Madison, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Electrical and Computer Engineering, University of Wisconsin-Madison, Madison, Unites States; 4Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, United States; 5The Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, United States Abstract How transcription factor dimerization impacts DNA-binding specificity is poorly understood. Guided by protein dimerization properties, we examined DNA binding specificities of 270 human bZIP pairs. DNA interactomes of 80 heterodimers and 22 homodimers revealed that 72% of heterodimer motifs correspond to conjoined half-sites preferred by partnering monomers. Remarkably, the remaining motifs are composed of variably-spaced half-sites (12%) or ‘emergent’ sites (16%) that cannot be readily inferred from half-site preferences of partnering monomers. -

Distinct Roles of Jun : Fos and Jun : ATF Dimers in Oncogenesis
Oncogene (2001) 20, 2453 ± 2464 ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00 www.nature.com/onc Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis Hans van Dam*,1 and Marc Castellazzi2 1Department of Molecular Cell Biology, Leiden University Medical Center, Sylvius Laboratories, PO Box 9503, 2300 RA Leiden, The Netherlands; 2Unite de Virologie Humaine, Institut National de la Sante et de la Recherche MeÂdicale (INSERM-U412), Ecole Normale SupeÂrieure, 46 alleÂe d'Italie, 69364 Lyon Cedex 07, France Jun : Fos and Jun : ATF complexes represent two classes dimers with emphasis on their roles in oncogenic of AP-1 dimers that (1) preferentially bind to either transformation in avian model systems. Previous heptameric or octameric AP-1 binding sites, and (2) are reviews on AP-1 and cell transformation include dierently regulated by cellular signaling pathways and references: (Angel and Karin, 1991; Wisdom, 1999; oncogene products. To discriminate between the func- Vogt, 1994; Karin et al., 1997; van Dam and van der tions of Jun : Fos, Jun: ATF and Jun : Jun, mutants were Eb, 1994; Hagmeyer et al., 1995). developed that restrict the ability of Jun to dimerize either to itself, or to Fos(-like) or ATF(-like) partners. Introduction of these mutants in chicken embryo Jun : Fos and Jun : ATF transcription factors: dimeric ®broblasts shows that Jun : Fra2 and Jun : ATF2 dimers complexes with variable composition and activities play distinct, complementary roles in in vitro oncogenesis by inducing either anchorage independence or growth AP-1 sub-units: members of the bZip protein family factor independence, respectively. -

Tgfβ-Regulated Gene Expression by Smads and Sp1/KLF-Like Transcription Factors in Cancer VOLKER ELLENRIEDER
ANTICANCER RESEARCH 28 : 1531-1540 (2008) Review TGFβ-regulated Gene Expression by Smads and Sp1/KLF-like Transcription Factors in Cancer VOLKER ELLENRIEDER Signal Transduction Laboratory, Internal Medicine, Department of Gastroenterology and Endocrinology, University of Marburg, Marburg, Germany Abstract. Transforming growth factor beta (TGF β) controls complex induces the canonical Smad signaling molecules which vital cellular functions through its ability to regulate gene then translocate into the nucleus to regulate transcription (2). The expression. TGFβ binding to its transmembrane receptor cellular response to TGF β can be extremely variable depending kinases initiates distinct intracellular signalling cascades on the cell type and the activation status of a cell at a given time. including the Smad signalling and transcription factors and also For instance, TGF β induces growth arrest and apoptosis in Smad-independent pathways. In normal epithelial cells, TGF β healthy epithelial cells, whereas it can also promote tumor stimulation induces a cytostatic program which includes the progression through stimulation of cell proliferation and the transcriptional repression of the c-Myc oncogene and the later induction of an epithelial-to-mesenchymal transition of tumor induction of the cell cycle inhibitors p15 INK4b and p21 Cip1 . cells (1, 3). In the last decade it has become clear that both the During carcinogenesis, however, many tumor cells lose their tumor suppressing and the tumor promoting functions of TGF β ability to respond to TGF β with growth inhibition, and instead, are primarily regulated on the level of gene expression through activate genes involved in cell proliferation, invasion and Smad-dependent and -independent mechanisms (1, 2, 4). -

Epigenetic Silencing of the Candidatetumor Suppressor Gene Per1 in Non ^ Small Cell Lung Cancer Sigal Gery,1Naoki Komatsu,1Norihiko Kawamata,1Carlw
Human Cancer Biology Epigenetic Silencing of the CandidateTumor Suppressor Gene Per1 in Non ^ Small Cell Lung Cancer Sigal Gery,1Naoki Komatsu,1Norihiko Kawamata,1CarlW. Miller,1Julian Desmond,1Renu K.Virk,1 Alberto Marchevsky,2 Robert Mckenna,3 Hirokuni Taguchi,4 and H. Phillip Koeffler1 Abstract Purpose: Epigenetic events are a critical factor contributing to cancer development.The purpose of this study was to identify tumor suppressor genes silenced by DNAmethylation and histone deacetylation in non ^ small cell lung cancer (NSCLC). Experimental Design: We used microarray analysis to screen for tumor suppressor genes. Results: We identified Per1, a core circadian gene, as a candidate tumor suppressor in lung cancer. Although Per1levels were high in normal lung, its expression was low in a large panel of NSCLC patient samples and cell lines. Forced expression of Per1in NSCLC cell lines led to signif- icant growth reduction and loss of clonogenic survival. Recent studies showed that epigenetic regulation, particularly histone H3 acetylation, is essential for circadian function. Using bisulfite sequencing and chromatin immunoprecipitation, we found that DNAhypermethylation and histone H3 acetylation are potential mechanisms for silencing Per1expression NSCLC. Conclusions:These results support the hypothesis that disruption of circadian rhythms plays an important role in lung tumorigenesis. Moreover, our findings suggest a novel link between circa- dian epigenetic regulation and cancer development. Lung caner is the leading cause of cancer-related death in the and early diagnosis. These recognitions have prompted United States (1, 2). Prevention, screening, and treatment of extensive research aimed at discovering silenced tumor sup- this cancer are all problematic, emphasizing the need for the pressors. -

1.1.4.1. Tumour Suppressor Genes
To my Mother and Father Learn from yesterday, live for today, hope for tomorrow. The important thing is not to stop questioning. Albert Einstein Inhibition of Tumourigenicity of Small Cell Lung Cancer by Simultaneous Suppression of Id1 and Id3 Expression Danqing Chen ABSTRACT Inhibitor of DNA binding (Id) proteins are a group of transcription factors belonging to the basic helix-loop-helix (bHLH) family and play a wide range of roles in differentiation, proliferation and cell cycle progression. Id proteins act as negative dominant regulators of other bHLH factors by making dimers to these factors to prevent them from binding to E-box of DNA and, hence, to inhibit transcription of target genes. In this work, we first established SCLC cell line N417-derived sublines expressing reduced levels of Id1 and Id3 by transfection of a single vector constructed to co-express two shRNAs simultaneously. Then we investigated the effect of either singly or jointly suppressed Id1 or Id3 on tumourigenicity of SCLC cells in vitro and in vivo. The molecular mechanisms involved in the functional roles of Id1 and Id3 were also assessed. Id1-suppressed cells and Id1 and Id3 double knockdown cells produced significant reductions in proliferation rate by more than 1.4- and 3.9-fold respectively when compared with the control. Soft agar assay showed the number of colonies produced by Id1-suppressed cells and Id1 and Id3 double knockdown cells were reduced by more than 13.7- and 233-fold respectively compared with the control. The suppression effect was also observed in the invasion assay which showed that Id1-suppressed cells and Id1 and Id3 double knockdown cells produced more than 1.7- and 4.6- fold reduction respectively in relative invasiveness. -

The Tumor Suppressor HHEX Inhibits Axon Growth When Prematurely Expressed in Developing Central Nervous System Neurons
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by epublications@Marquette Marquette University e-Publications@Marquette Biological Sciences Faculty Research and Biological Sciences, Department of Publications 9-1-2015 The umorT Suppressor HHEX Inhibits Axon Growth when Prematurely Expressed in Developing Central Nervous System Neurons Matthew .T Simpson Marquette University Ishwariya Venkatesh Marquette University Ben L. Callif Marquette University Laura K. Thiel Marquette University Denise M. Coley Marquette University See next page for additional authors Accepted version. Molecular and Cellular Neuroscience, Vol 68 )September 2015): 272-283. DOI. © 2015 Elsevier Inc. Used with permission. NOTICE: this is the author’s version of a work that was accepted for publication in Molecular and Cellular Neuroscience. Changes resulting from the publishing process, such as peer review, editing, corrections, structural formatting, and other quality control mechanisms may not be reflected in this document. Changes may have been made to this work since it was submitted for publication. A definitive version was subsequently published in Molecular and Cellular Neuroscience, Vol 68 )September 2015): 272-283. DOI. Authors Matthew T. Simpson, Ishwariya Venkatesh, Ben L. Callif, Laura K. Thiel, Denise M. Coley, Kristen N. Winsor, Zimei Wang, Audra A. Kramer, Jessica K. Lerch, and Murray G. Blackmore This article is available at e-Publications@Marquette: https://epublications.marquette.edu/bio_fac/515 NOT THE PUBLISHED VERSION; this is the author’s final, peer-reviewed manuscript. The published version may be accessed by following the link in the citation at the bottom of the page. The Tumor Suppressor HHEX Inhibits Axon Growth When Prematurely Expressed in Developing Central Nervous System Neurons Matthew T. -
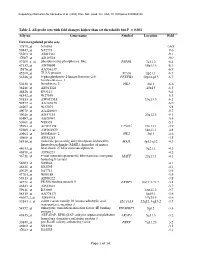
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Role of Activating Transcription Factor 3 in the Synthesis of Latency
Role of activating transcription factor 3 in the synthesis PNAS PLUS of latency-associated transcript and maintenance of herpes simplex virus 1 in latent state in ganglia Minfeng Shu, Te Du, Grace Zhou1, and Bernard Roizman2 Marjorie B. Kovler Viral Oncology Laboratories, The University of Chicago, Chicago IL 60637 Contributed by Bernard Roizman, August 4, 2015 (sent for review July 17, 2015) A key property of herpes simplex viruses (HSVs) is their ability to rapidly deplete NGF or in medium containing both NGF and establish latent infection in sensory or autonomic ganglia and to epithelial growth factor (EGF) (9). In the former medium, ex- reactivate on physical, hormonal, or emotional stress. In latently pression of viral genes begins at ∼5 h after excision of the gan- infected ganglia, HSVs express a long noncoding RNA, a latency- glia. In medium containing NGF and EGF, the virus remains associated transcript (LAT), which plays a key role in maintaining silent for ∼18–24 h. Studies on this model revealed that in latently infected neurons, but not viral proteins. To investigate the ganglia maintained in medium containing NGF plus EGF, the events leading to reactivation, we examined the use of ganglionic virus is maintained in a dynamic equilibrium that is disrupted organ cultures that enable rapid reactivation in medium contain- by inhibitors of HDAC-1 and -4, the PI3K-AKT-mTOR and STAT3 ing antibody to nerve growth factor (NGF) or delayed reactivation pathways, or proapoptotic drugs (10, 11). Reactivation in medium in medium containing NGF and epidermal growth factor (EGF). containing anti-NGF antibody is inhibited by the expression of Here we report the discovery that activating transcription factor 3 REST or by a p300/CBP inhibitor (11, 12). -

Pathways of NF-Κb Activation
Biochemical Journal (2021) 478 2619–2664 https://doi.org/10.1042/BCJ20210139 Review Article Inhibitory feedback control of NF-κB signalling in health and disease Jack A. Prescott*, Jennifer P. Mitchell and Simon J. Cook Signalling Programme, The Babraham Institute, Babraham Research Campus, Cambridge CB22 3AT, U.K. Correspondence: Simon Cook ([email protected]) or Jennifer Mitchell ( [email protected]) Cells must adapt to changes in their environment to maintain cell, tissue and organismal integrity in the face of mechanical, chemical or microbiological stress. Nuclear factor-κB (NF-κB) is one of the most important transcription factors that controls inducible gene expression as cells attempt to restore homeostasis. It plays critical roles in the immune system, from acute inflammation to the development of secondary lymphoid organs, and also has roles in cell survival, proliferation and differentiation. Given its role in such critical processes, NF-κB signalling must be subject to strict spatiotemporal control to ensure measured and context-specific cellular responses. Indeed, deregulation of NF-κB signal- ling can result in debilitating and even lethal inflammation and also underpins some forms of cancer. In this review, we describe the homeostatic feedback mechanisms that limit and ‘re-set’ inducible activation of NF-κB. We first describe the key components of the signalling pathways leading to activation of NF-κB, including the prominent role of protein phosphorylation and protein ubiquitylation, before briefly introducing the key fea- tures of feedback control mechanisms. We then describe the array of negative feedback loops targeting different components of the NF-κB signalling cascade including controls at the receptor level, post-receptor signalosome complexes, direct regulation of the crit- ical ‘inhibitor of κB kinases’ (IKKs) and inhibitory feedforward regulation of NF-κB- dependent transcriptional responses. -

Single-Cell Transcriptome Profiling an Adult Human Cell Atlas of 15 Major Organs
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.996975; this version posted March 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Single-cell transcriptome profiling an adult human cell atlas of 15 major organs Shuai He1,2,3,4,#, Lin-he Wang2,3,4,#, Yang Liu1 ,#, Yi-qi Li1, Haitian Chen2,3,4, Jinghong Xu2,3,4, Wan Peng1, Guo-Wang Lin1, Pan-Pan Wei1, Bo Li5,6, Xiaojun Xia1, Dan Wang1, Jin-Xin Bei1,7,*, Xiaoshun He2,3,4,*, Zhiyong Guo2,3,4,* 1Sun Yat-sen University Cancer Centre, State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine, Guangzhou 510060, P. R. China. 2Organ Transplant Center, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China 3Guangdong Provincial Key Laboratory of Organ Donation and Transplant Immunology, Guangzhou 510080, China 4Guangdong Provincial International Cooperation Base of Science and Technology (Organ Transplantation), Guangzhou 510080, China 5Department of Biochemistry and Molecular Biology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, P. R. China 6RNA Biomedical Institute, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 510120, P. R. China 7Centre for Precision Medicine, Sun Yat-sen University, Guangzhou 510080, P. R. China. Email addresses of authors: Shuai He, [email protected]; Lin-he Wang, [email protected]; Yang Liu, [email protected]; Yi-qi Li, [email protected]; Haitian Chen, [email protected]; Jinghong Xu, [email protected]; Wan Peng, [email protected]; Guo-Wang Lin, [email protected]; Pan-Pan Wei, [email protected]; Bo Li, [email protected]; Xiaojun Xia, [email protected]; Dan Wang, [email protected]; Jin-Xin Bei, [email protected]; Xiaoshun He, [email protected]; Zhiyong Guo, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.996975; this version posted March 20, 2020. -

A Promoter Polymorphism in the Per3 Gene Is Associated with Alcohol and Stress Response
Citation: Transl Psychiatry (2012) 2, e73, doi:10.1038/tp.2011.71 & 2012 Macmillan Publishers Limited All rights reserved 2158-3188/12 www.nature.com/tp A promoter polymorphism in the Per3 gene is associated with alcohol and stress response X Wang1, K Mozhui1,ZLi1, MK Mulligan1, JF Ingels1, X Zhou1, RT Hori2, H Chen3, MN Cook4, RW Williams1 and L Lu1,5 The period homolog genes Per1, Per2 and Per3 are important components of the circadian clock system. In addition to their role in maintaining circadian rhythm, these genes have been linked to mood disorders, stress response and vulnerability to addiction and alcoholism. In this study, we combined high-resolution sequence analysis and quantitative trait locus (QTL) mapping of gene expression and behavioral traits to identify Per3 as a compelling candidate for the interaction between circadian rhythm, alcohol and stress response. In the BXD family of mouse strains, sequence variants in Per3 have marked effects on steady-state mRNA and protein levels. As a result, the transcript maps as a cis-acting expression QTL (eQTL). We found that an insertion/deletion (indel) variant in a putative stress response element in the promoter region of Per3 causes local control of transcript abundance. This indel results in differences in protein binding affinities between the two alleles through the Nrf2 transcriptional activator. Variation in Per3 is also associated with downstream differences in the expression of genes involved in circadian rhythm, alcohol, stress response and schizophrenia. We found that the Per3 locus is linked to stress/anxiety traits, and that the basal expression of Per3 is also correlated with several anxiety and addiction-related phenotypes.