Cell Cycle Arrest Through Indirect Transcriptional Repression by P53: I Have a DREAM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CDC25B Mediates Rapamycin-Induced Oncogenic Responses in Cancer Cells
Published OnlineFirst March 10, 2009; DOI: 10.1158/0008-5472.CAN-08-3222 Research Article CDC25B Mediates Rapamycin-Induced Oncogenic Responses in Cancer Cells Run-qiang Chen,1 Qing-kai Yang,1 Bing-wen Lu,2 Wei Yi,1 Greg Cantin,2 Yan-ling Chen,1 Colleen Fearns,3 John R. Yates III,2 and Jiing-Dwan Lee1 Departments of 1Immunology and Microbial Science, 2Chemical Physiology, and 3Chemistry, The Scripps Research Institute, La Jolla, California Abstract expression of PTEN, increased PI3K activity, and increased expression or activation of AKT in advanced prostate cancer Because the mammalian target of rapamycin (mTOR) pathway (8–10). These aberrations also are indicators of a poor prognosis is commonly deregulated in human cancer, mTOR inhibitors, for prostate cancer patients (11, 12). More importantly, long-term rapamycin and its derivatives, are being actively tested in androgen deprivation treatment for prostate cancer patients that cancer clinical trials. Clinical updates indicate that the reinforces the PI3K/AKT pathway also up-regulates mTOR anticancer effect of these drugs is limited, perhaps due to activation in prostate tumor (9, 10). These abovementioned rapamycin-dependent induction of oncogenic cascades by an experimental and clinical data lead to the supposition that mTOR as yet unclear mechanism. As such, we investigated rapamy- inhibitors (rapamycin and its derivatives) should be effective in cin-dependent phosphoproteomics and discovered that 250 treating human cancer. Unfortunately, recent clinical data indicates phosphosites in 161 cellular proteins were sensitive to that rapamycin shows therapeutic potential in only few types of rapamycin. Among these, rapamycin regulated four kinases human cancer: endometrial carcinoma, renal cell carcinoma, and and four phosphatases. -

Identification of Hub Genes Involved in the Occurrence and Development of Hepatocellular Carcinoma Via Bioinformatics Analysis
ONCOLOGY LETTERS 20: 1695-1708, 2020 Identification of hub genes involved in the occurrence and development of hepatocellular carcinoma via bioinformatics analysis NINGNING MI1,2*, JIE CAO1-6*, JINDUO ZHANG2-5, WENKANG FU1-5, CHONGFEI HUANG1-5, LONG GAO1-5, PING YUE2-5, BING BAI2-5, YANYAN LIN1-5, WENBO MENG1-5 and XUN LI4,5,7 1The First Clinical Medical School, Lanzhou University; 2Department of Special Minimally Invasive Surgery, The First Hospital of Lanzhou University; 3Institute of Genetics, School of Basic Medical Sciences, Lanzhou University; 4Gansu Province Institute of Hepatopancreatobiliary; 5Gansu Province Key Laboratory Biotherapy and Regenerative Medicine; 6Laboratory Department; 7The Fifth Department of General Surgery, The First Hospital of Lanzhou University, Lanzhou, Gansu 730000, P.R. China Received September 28, 2019; Accepted May 7, 2020 DOI: 10.3892/ol.2020.11752 Abstract. Hepatocellular carcinoma (HCC) is a heterogeneous cyclin B1 (CCNB1), cell-division cycle protein 20 (CDC20), malignancy, which is a major cause of cancer morbidity and cyclin-dependent kinase 1, BUB1 mitotic checkpoint serine/ mortality worldwide. Thus, the aim of the present study was threonine kinase β (BUB1B), cyclin A2, nucleolar and spindle to identify the hub genes and underlying pathways of HCC associated protein 1, ubiquitin‑conjugating enzyme E2 C via bioinformatics analyses. The present study screened three (UBE2C) and ZW10 interactor. Furthermore, upregulated datasets, including GSE112790, GSE84402 and GSE74656 CCNB1, CDC20, BUB1B and UBE2C expression levels from the Gene Expression Omnibus (GEO) database, and indicated worse disease-free and overall survival. Moreover, downloaded the RNA-sequencing of HCC from The Cancer a meta-analysis of tumor and healthy tissues in the Oncomine Genome Atlas (TCGA) database. -
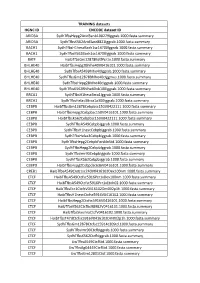
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Biallelic TRIP13 Mutations Predispose to Wilms Tumor and Chromosome Missegregation
Europe PMC Funders Group Author Manuscript Nat Genet. Author manuscript; available in PMC 2017 July 01. Published in final edited form as: Nat Genet. 2017 July ; 49(7): 1148–1151. doi:10.1038/ng.3883. Europe PMC Funders Author Manuscripts Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation Shawn Yost#1, Bas de Wolf#2, Sandra Hanks#1, Anna Zachariou1, Chiara Marcozzi3,4, Matthew Clarke1, Richarda de Voer2, Banafsheh Etemad2, Esther Uijttewaal2, Emma Ramsay1, Harriet Wylie1, Anna Elliott1, Susan Picton5, Audrey Smith6, Sarah Smithson7, Sheila Seal1, Elise Ruark1, Gunnar Houge8, Jonathan Pines3,4, Geert J.P.L. Kops2,9,10,+, and Nazneen Rahman1,11,+ 1Division of Genetics and Epidemiology, Institute of Cancer Research, 15 Cotswold Road, London, SM2 5NG, UK 2Hubrecht Institute – KNAW (Royal Netherlands Academy of Arts and Sciences), Uppsalalaan 8, 3584 CT Utrecht, The Netherlands 3The Gurdon Institute and Department of Zoology, University of Cambridge, Cambridge CB2 1QN, UK 4Division of Cancer Biology, The Institute of Cancer Research, 237 Fulham Road, London SW3 6JB, UK 5Children's and Adolescent Oncology and Haematology Unit, Leeds General Infirmary, Leeds, LS1 3EX, UK 6Yorkshire Regional Clinical Genetics Service, Chapel Allerton Hospital, Chapeltown Road, Leeds, LS7 4SA, UK 7Clinical Genetics Service, St Michael's Hospital, Southwell Street, Bristol, BS2 8EG, UK 8Center for Medical Genetics, Haukeland University Hospital, N-5021 Bergen, Norway 9Cancer Genomics Netherlands, Utrecht, The Netherlands 10Center for Molecular 11 Europe PMC Funders Author Manuscripts Medicine, University Medical Center Utrecht, 3584 CG, Utrecht, The Netherlands Cancer Genetics Unit, Royal Marsden NHS Foundation Trust, London, UK SM2 5PT, UK # These authors contributed equally to this work. -

The Architecture of a Eukaryotic Replisome
The Architecture of a Eukaryotic Replisome Jingchuan Sun1,2, Yi Shi3, Roxana E. Georgescu3,4, Zuanning Yuan1,2, Brian T. Chait3, Huilin Li*1,2, Michael E. O’Donnell*3,4 1 Biosciences Department, Brookhaven National Laboratory, Upton, New York, USA 2 Department of Biochemistry & Cell Biology, Stony Brook University, Stony Brook, New York, USA. 3 The Rockefeller University, 1230 York Avenue, New York, New York, USA. 4 Howard Hughes Medical Institute *Correspondence and requests for materials should be addressed to M.O.D. ([email protected]) or H.L. ([email protected]) ABSTRACT At the eukaryotic DNA replication fork, it is widely believed that the Cdc45-Mcm2-7-GINS (CMG) helicase leads the way in front to unwind DNA, and that DNA polymerases (Pol) trail behind the helicase. Here we use single particle electron microscopy to directly image a replisome. Contrary to expectations, the leading strand Pol ε is positioned ahead of CMG helicase, while Ctf4 and the lagging strand Pol α-primase (Pol α) are behind the helicase. This unexpected architecture indicates that the leading strand DNA travels a long distance before reaching Pol ε, it first threads through the Mcm2-7 ring, then makes a U-turn at the bottom to reach Pol ε at the top of CMG. Our work reveals an unexpected configuration of the eukaryotic replisome, suggests possible reasons for this architecture, and provides a basis for further structural and biochemical replisome studies. INTRODUCTION DNA is replicated by a multi-protein machinery referred to as a replisome 1,2. Replisomes contain a helicase to unwind DNA, DNA polymerases that synthesize the leading and lagging strands, and a primase that makes short primed sites to initiate DNA synthesis on both strands. -

Orpl, a Member of the Cdcl8/Cdc6 Family of S-Phase Regulators, Is Homologous to a Component of the Origin Recognition Complex M
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 12475-12479, December 1995 Genetics Orpl, a member of the Cdcl8/Cdc6 family of S-phase regulators, is homologous to a component of the origin recognition complex M. MuzI-FALCONI AND THOMAS J. KELLY Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, Baltimore, MD 21205 Contributed by Thomas J. Kelly, September 11, 1995 ABSTRACT cdc18+ of Schizosaccharomyces pombe is a is the cdcJ8+ gene (1, 15). Expression of cdcJ8+ from a periodically expressed gene that is required for entry into S heterologous promoter is sufficient to rescue the lethality of a phase and for the coordination of S phase with mitosis. cdc18+ conditional temperature-sensitive (ts) cdc O's mutant. The is related to the Saccharomyces cerevisiae gene CDC6, which has cdcJ8+ gene product, a 65-kDa protein, is essential for the also been implicated in the control of DNA replication. We GI/S transition. Moreover, p65cdclS is a highly labile protein have identified a new Sch. pombe gene, orpl1, that encodes an whose expression is confined to a narrow window at the G,/S 80-kDa protein with amino acid sequence motifs conserved in boundary (unpublished data). These properties are consistent the Cdc18 and Cdc6 proteins. Genetic analysis indicates that with the hypothesis that Cdc18 may play an important role in orpi + is essential for viability. Germinating spores lacking the regulating the initiation of DNA replication at S phase. The orpl + gene are capable of undergoing one or more rounds of Cdc18 protein is homologous to the budding yeast Cdc6 DNA replication but fail to progress further, arresting as long protein, which may have a similar function (16). -

Calpain Inhibitors Prevent Nitric Oxide-Triggered Excitotoxic Apoptosis
Calpain inhibitors prevent nitric oxide- triggered excitotoxic apoptosis Christiane Volbracht,1,2 Eugenio Fava,1,3 Marcel Leist1,4 and Pierluigi Nicotera1,3,CA 1Molecular Toxicology, University of Konstanz, Konstanz, Germany; 2Institute of Molecular and Cell Biology, Singapore 117609, Singapore; 3MRC Toxicology Unit, University of Leicester, PO Box 138, Lancaster Road, Leicester LE1 9HN; 4Department of Neurobiology, H. Lundbeck A/S, 2500 Valby, Denmark CACorresponding Author The pathogenesis of some neurodegenerative disorders has potential, chromatin breakdown, and subsequent death of been linked to excitotoxicity, excess generation of nitric oxide cerebellar granule neurons exposed to NO donors (S-nitroso- (NO) and apoptosis. Here, we used a model of NO-triggered L-glutathione, S-nitroso-N-acetyl-D,L-penicillamine, and diethy- neuronal apoptosis that was strictly dependent on autocrine lamino-diazenolate-2-oxide). Since inhibitors did not interfere 2 NMDA receptor (NMDA-R) activation and intracellular Ca with NMDA-R activation, we suggest that block of calpains increase. We investigated the ef®ciency and potentially bene- blunts NO-triggered neuronal apoptosis by stopping the ®cial effects of calpain inhibition. Three calpain inhibitors that cascade downstream of primary autocrine excitotoxic events. prevented intracellular fodrin proteolysis also blocked apopto- NeuroReport 12:3645±3648 & 2001 Lippincott Williams & tic features such as decrease in mitochondrial membrane Wilkins. Key words: Apoptosis; Calpains; Excitotoxicity; Mitochondria; Nitric oxide INTRODUCTION MATERIALS AND METHODS Massive generation of the pleiotropic messenger molecule Cell culture: Murine CGC were isolated from 8-day-old nitric oxide (NO) has been implicated in many neuro- speci®c pathogen free BALB/e mice obtained from the pathological conditions including ischemia [1]. -

Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement
School of Natural Sciences and Mathematics Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement UT Dallas Author(s): Michael Q. Zhang Rights: CC BY 4.0 (Attribution) ©2017 The Authors Citation: Liu, Zehua, Huazhe Lou, Kaikun Xie, Hao Wang, et al. 2017. "Reconstructing cell cycle pseudo time-series via single-cell transcriptome data." Nature Communications 8, doi:10.1038/s41467-017-00039-z This document is being made freely available by the Eugene McDermott Library of the University of Texas at Dallas with permission of the copyright owner. All rights are reserved under United States copyright law unless specified otherwise. File name: Supplementary Information Description: Supplementary figures, supplementary tables, supplementary notes, supplementary methods and supplementary references. CCNE1 CCNE1 CCNE1 CCNE1 36 40 32 34 32 35 30 32 28 30 30 28 28 26 24 25 Normalized Expression Normalized Expression Normalized Expression Normalized Expression 26 G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage CCNE1 CCNE1 CCNE1 CCNE1 40 32 40 40 35 30 38 30 30 28 36 25 26 20 20 34 Normalized Expression Normalized Expression Normalized Expression 24 Normalized Expression G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Supplementary Figure 1 | High stochasticity of single-cell gene expression means, as demonstrated by relative expression levels of gene Ccne1 using the mESC-SMARTer data. For every panel, 20 sample cells were randomly selected for each of the three stages, followed by plotting the mean expression levels at each stage. -

Production of Circularly Permuted Caspase-2 for Affinity Fusion-Tag
biomolecules Article Production of Circularly Permuted Caspase-2 for Affinity Fusion-Tag Removal: Cloning, Expression in Escherichia coli, Purification, and Characterization 1,2, 1,2, , 1,3 1,3 Monika Cserjan-Puschmann y , Nico Lingg * y , Petra Engele , Christina Kröß , Julian Loibl 1, Andreas Fischer 1, Florian Bacher 1, Anna-Carina Frank 1,2, Christoph Öhlknecht 1,4,Cécile Brocard 5, Chris Oostenbrink 1,4 , Matthias Berkemeyer 5, Rainer Schneider 1,3, Gerald Striedner 1,2 and Alois Jungbauer 1,2,* 1 ACIB-Austrian Centre of Industrial Biotechnology, Muthgasse 18, 1190 Vienna, Austria; [email protected] (M.C.-P.); [email protected] (P.E.); [email protected] (C.K.); [email protected] (J.L.); andreas.fi[email protected] (A.F.); fl[email protected] (F.B.); [email protected] (A.-C.F.); [email protected] (C.Ö.); [email protected] (C.O.); [email protected] (R.S.); [email protected] (G.S.) 2 Department of Biotechnology, University of Natural Resources and Life Sciences, Vienna, Muthgasse 18, 1190 Vienna, Austria 3 Institute of Biochemistry, Center for Molecular Biosciences Innsbruck (CMBI), University of Innsbruck, 6020 Innsbruck, Austria 4 Institute of Molecular Modeling and Simulation, University of Natural Resources and Life Sciences, Vienna, Muthgasse 18, 1190 Vienna, Austria 5 Biopharma Process Science Austria, Boehringer Ingelheim RCV GmbH & Co KG, Dr. Boehringer-Gasse 5-11, 1121 Vienna, Austria; [email protected] (C.B.); [email protected] (M.B.) * Correspondence: [email protected] (N.L.); [email protected] (A.J.) These authors have contributed equally to the manuscript. -

Anillin Is a Prognostic Factor and Is Correlated with Genovariation in Pancreatic Cancer Based on Databases Analysis
ONCOLOGY LETTERS 21: 107, 2021 Anillin is a prognostic factor and is correlated with genovariation in pancreatic cancer based on databases analysis YUANHUA NIE, ZHIQIANG ZHAO, MINXUE CHEN, FULIN MA, YONG FAN, YINGXIN KANG, BOXIONG KANG and CHEN WANG Department of General Surgery, Lanzhou University Second Hospital, Lanzhou, Gansu 730030, P.R. China Received March 12, 2020; Accepted October 8, 2020 DOI: 10.3892/ol.2020.12368 Abstract. Pancreatic cancer has a low survival rate globally. PC pathways were associated with low expression of ANLN. Anillin (ANLN) is involved in the pathogenesis of pancreatic Overall, ANLN is more highly expressed in PC compared cancer (PC). The present study used databases and reverse with in normal tissue, and is associated with poor differen‑ transcription‑quantitative PCR to investigate the association tiation. The expression of ANLN may be a novel prognostic between ANLN expression, clinical variables and the survival marker of poor survival. Finally, ANLN exert its functions in rate of patients with pancreatic cancer. Gene expression PC through the p53, cell cycle, DNA replication, mismatch of ANLN in normal and cancer tissues was analyzed using repair and nucleotide excision repair and pathways. data from The Cancer Genome Atlas, Oncomine and Gene Expression database of Normal and Tumor tissues 2 and Introduction ANOVA, and the association between ANLN mRNA expres‑ sion and ANLN genovariation was analyzed using cBioPortal. Pancreatic cancer (PC) is a major public health problem The association between ANLN expression and the survival, and is the eleventh most common cancer in the world, with clinical, pathological and prognostic characteristics of PC 458,918 new cases and 432,242 deaths in 2018 (1). -

Screening and Identification of Hub Genes in Bladder Cancer by Bioinformatics Analysis and KIF11 Is a Potential Prognostic Biomarker
ONCOLOGY LETTERS 21: 205, 2021 Screening and identification of hub genes in bladder cancer by bioinformatics analysis and KIF11 is a potential prognostic biomarker XIAO‑CONG MO1,2*, ZI‑TONG ZHANG1,3*, MENG‑JIA SONG1,2, ZI‑QI ZHOU1,2, JIAN‑XIONG ZENG1,2, YU‑FEI DU1,2, FENG‑ZE SUN1,2, JIE‑YING YANG1,2, JUN‑YI HE1,2, YUE HUANG1,2, JIAN‑CHUAN XIA1,2 and DE‑SHENG WENG1,2 1State Key Laboratory of Oncology in South China, Collaborative Innovation Centre for Cancer Medicine; 2Department of Biotherapy, Sun Yat‑Sen University Cancer Center; 3Department of Radiation Oncology, Sun Yat‑Sen University Cancer Center, Guangzhou, Guangdong 510060, P.R. China Received July 31, 2020; Accepted December 18, 2020 DOI: 10.3892/ol.2021.12466 Abstract. Bladder cancer (BC) is the ninth most common immunohistochemistry and western blotting. In summary, lethal malignancy worldwide. Great efforts have been devoted KIF11 was significantly upregulated in BC and might act as to clarify the pathogenesis of BC, but the underlying molecular a potential prognostic biomarker. The present identification mechanisms remain unclear. To screen for the genes associated of DEGs and hub genes in BC may provide novel insight for with the progression and carcinogenesis of BC, three datasets investigating the molecular mechanisms of BC. were obtained from the Gene Expression Omnibus. A total of 37 tumor and 16 non‑cancerous samples were analyzed to Introduction identify differentially expressed genes (DEGs). Subsequently, 141 genes were identified, including 55 upregulated and Bladder cancer (BC) is the ninth most common malignancy 86 downregulated genes. The protein‑protein interaction worldwide with substantial morbidity and mortality. -

Genetic Drivers of Pancreatic Islet Function
| INVESTIGATION Genetic Drivers of Pancreatic Islet Function Mark P. Keller,*,1 Daniel M. Gatti,†,1 Kathryn L. Schueler,* Mary E. Rabaglia,* Donnie S. Stapleton,* Petr Simecek,† Matthew Vincent,† Sadie Allen,‡ Aimee Teo Broman,§ Rhonda Bacher,§ Christina Kendziorski,§ Karl W. Broman,§ Brian S. Yandell,** Gary A. Churchill,†,2 and Alan D. Attie*,2 *Department of Biochemistry, §Department of Biostatistics and Medical Informatics, and **Department of Horticulture, University of Wisconsin–Madison, Wisconsin 53706-1544, †The Jackson Laboratory, Bar Harbor, Maine 06409, and ‡Maine School of Science and Mathematics, Limestone, Maine 06409, ORCID IDs: 0000-0002-7405-5552 (M.P.K.); 0000-0002-4914-6671 (K.W.B.); 0000-0001-9190-9284 (G.A.C.); 0000-0002-0568-2261 (A.D.A.) ABSTRACT The majority of gene loci that have been associated with type 2 diabetes play a role in pancreatic islet function. To evaluate the role of islet gene expression in the etiology of diabetes, we sensitized a genetically diverse mouse population with a Western diet high in fat (45% kcal) and sucrose (34%) and carried out genome-wide association mapping of diabetes-related phenotypes. We quantified mRNA abundance in the islets and identified 18,820 expression QTL. We applied mediation analysis to identify candidate causal driver genes at loci that affect the abundance of numerous transcripts. These include two genes previously associated with monogenic diabetes (PDX1 and HNF4A), as well as three genes with nominal association with diabetes-related traits in humans (FAM83E, IL6ST, and SAT2). We grouped transcripts into gene modules and mapped regulatory loci for modules enriched with transcripts specific for a-cells, and another specific for d-cells.