Development and Applications of Novel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Theoretical Study of Sarin Adsorption On
Chemical Physics Letters 738 (2020) 136816 Contents lists available at ScienceDirect Chemical Physics Letters journal homepage: www.elsevier.com/locate/cplett Research paper Theoretical study of sarin adsorption on (12,0) boron nitride nanotube doped with silicon atoms T ⁎ ⁎ Jeziel Rodrigues dos Santosa, , Elson Longo da Silvab, Osmair Vital de Oliveirac, , José Divino dos Santosa a Universidade Estadual de Goiás, Campus Anápolis, CEP: 75.132-903 GO, Brazil b INCTMN, LIEC, Departamento de Química da Universidade Federal de São Carlos, CEP: 13.565-905 São Carlos, SP, Brazil c Instituto Federal de Educação, Ciência e Tecnologia de São Paulo, Campus Catanduva, CEP: 15.808-305 Catanduva, SP, Brazil HIGHLIGHTS • DFT method was used to study the adsorption of nerve agent sarin by BNNT. • Electronic properties of pristine BNNT are improved by Si impurity atoms. • The adsorption of sarin by Si-doped BNNT is highest favorable than the pure BNNT. • Si-doped BNNT can be a new gas sensor for sarin gas detection and its derivatives. ARTICLE INFO ABSTRACT Keywords: Sarin gas is one of the most lethal nerve agent used in chemical warfare, which its detection is import to prevent Nerve agent sarin a chemical attack and to identify a contamination area. Herein, density functional theory was used to investigate Gas sensor the (12,0) boron nitride nanotube (BNNT) and Si–doped BNNT as possible candidates to sarin detection. The Si- Boron nitride nanotube atoms doped improve the electronic properties of nanotubes by altering the electrostatic potential, HOMO and DFT LUMO energies. Based in the adsorption energies and the conductivity increased to ~33 and 350%, respectively, for Si- and 2Si-BNNT imply that they can be used for sarin detection. -
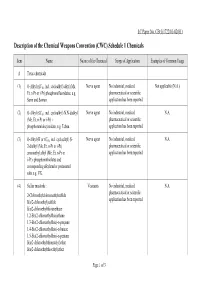
Description of the Chemical Weapons Convention (CWC) Schedule 1 Chemicals
LC Paper No. CB(1)1722/01-02(01) Description of the Chemical Weapons Convention (CWC) Schedule 1 Chemicals Item Name Nature of the Chemical Scope of Application Examples of Common Usage A Toxic chemicals (1) O-Alkyl (≤C10, incl. cycloalkyl) alkyl (Me, Nerve agent No industrial, medical, Not applicable (N.A.) Et, n-Pr or i-Pr) phosphonofluoridates, e.g. pharmaceutical or scientific Sarin and Soman. application has been reported. (2) O-Alkyl (≤C10, incl. cycloalkyl) N,N-dialkyl Nerve agent No industrial, medical, N.A. (Me, Et, n-Pr or i-Pr) - pharmaceutical or scientific phosphoramidocyanidate, e.g. Tabun. application has been reported. (3) O-Alkyl (H or ≤C10, incl. cycloalkyl) S- Nerve agent No industrial, medical, N.A. 2-dialkyl (Me, Et, n-Pr or i-Pr) pharmaceutical or scientific aminoethyl alkyl (Me, Et, n-Pr or application has been reported. i-Pr)- phosphonothiolates and corresponding alkylated or protonated salts e.g. VX. (4) Sulfur mustards : Vesicants No industrial, medical, N.A. pharmaceutical or scientific 2-Chloroethylchloromethylsulfide application has been reported. Bis(2-chloroethyl)sulfide Bis(2-chloroethylthio)methane 1,2-Bis(2-chloroethylthio)ethane 1,3-Bis(2-chloroethylthio)-n-propane 1,4-Bis(2-chloroethylthio)-n-butane 1,5-Bis(2-chloroethylthio)-n-pentane Bis(2-chloroethylthiomethyl)ether Bis(2-chloroethylthioethyl)ether Page 1 of 3 Item Name Nature of the Chemical Scope of Application Examples of Common Usage (5) Lewisites : Vesicants No industrial, medical, N.A. pharmaceutical or scientific Lewisite 1 : 2-Chlorovinyldichloroarsine application has been reported. Lewisite 2 : Bis(2-chlorovinyl)chloroarsine Lewisite 3 : Tris(2-chlorovinyl)arsine (6) Nitrogen mustards : Vesicants The chemical has medical Only HN2 has been reported to application. -

"The Science for Diplomats" Annex on Chemicals
ORGANISATION FOR THE PROHIBITION OF CHEMICAL WEAPONS "THE SCIENCE FOR DIPLOMATS" ANNEX ON CHEMICALS A user friendly and scientifically annotated version of the Chemical Weapons Convention Annex on Chemicals OPCW THE “SCIENCE FOR DIPLOMATS” ANNEX ON CHEMICALS A user friendly and scientifically annotated version of the Chemical Weapons Convention Annex on Chemicals1 CONTENTS A. GUIDELINES FOR SCHEDULES OF CHEMICALS B. VISUALISING AND READING MOLECULAR STRUCTURES C. SCHEDULES OF CHEMICALS D. RIOT CONTROL AGENTS 1 An official version of the Annex on Chemicals can be obtained from the OPCW public website, www.opcw.org/chemical-weapons-convention/annexes/annex-chemicals/annex-chemicals. Version 3.0 – 10 March 2019 A. GUIDELINES FOR SCHEDULES OF CHEMICALS Guidelines for Schedule 1 1. The following criteria shall be taken into account in considering whether a toxic chemical or precursor should be included in Schedule 1: (a) It has been developed, produced, stockpiled or used as a chemical weapon as defined in Article II; (b) It poses otherwise a high risk to the object and purpose of this Convention by virtue of its high potential for use in activities prohibited under this Convention because one or more of the following conditions are met: (i) It possesses a chemical structure closely related to that of other toxic chemicals listed in Schedule 1, and has, or can be expected to have, comparable properties; (ii) It possesses such lethal or incapacitating toxicity as well as other properties that would enable it to be used as a chemical weapon; (iii) It may be used as a precursor in the final single technological stage of production of a toxic chemical listed in Schedule 1, regardless of whether this stage takes place in facilities, in munitions or elsewhere; (c) It has little or no use for purposes not prohibited under this Convention. -

Table of Contents
UNIVERSITY OF MISSOURI-KANSAS CITY CHEMICAL MANAGEMENT PLAN Revised May 2016 UMKC CHEMICAL MANAGEMENT PLAN This document constitutes the Chemical Management Plan (CMP) for the University of Missouri-Kansas City (UMKC). It was developed by the Environmental Health and Safety Department (EHS), to ensure the safe and proper use of hazardous and non- hazardous chemicals and to comply with applicable governmental regulations addressing the disposal of these chemicals. In addition, it was developed to foster waste minimization, and to provide the faculty and the staff with a management program to reduce the potential for accidents involving hazardous chemicals and/or wastes. Elements of the CMP include: a. a procedure for identifying potential or actual hazardous chemicals or wastes b. a procedure for periodic reexamination of those hazardous chemicals or wastes identified by the procedure in (a.) above as well as a systematic method for identification and evaluation of any new potential or actual hazardous chemicals or wastes c. procedures for labeling, and inventorying hazardous chemicals or wastes d. a procedure for identification and training of personnel directly responsible for ensuring that (a.), (b.), and (c.) are implemented e. a procedure for monitoring, recording, and reporting compliance with the CMP f. a procedure by which information generated by the CMP is provided to the persons performing waste analyses Each element is addressed as part of the complete CMP in the following paragraphs. 4 Table of Contents 1 Definitions 7 2 Identification -

Acetylcholinesterase: the “Hub” for Neurodegenerative Diseases And
Review biomolecules Acetylcholinesterase: The “Hub” for NeurodegenerativeReview Diseases and Chemical Weapons Acetylcholinesterase: The “Hub” for Convention Neurodegenerative Diseases and Chemical WeaponsSamir F. de A. Cavalcante Convention 1,2,3,*, Alessandro B. C. Simas 2,*, Marcos C. Barcellos 1, Victor G. M. de Oliveira 1, Roberto B. Sousa 1, Paulo A. de M. Cabral 1 and Kamil Kuča 3,*and Tanos C. C. França 3,4,* Samir F. de A. Cavalcante 1,2,3,* , Alessandro B. C. Simas 2,*, Marcos C. Barcellos 1, Victor1 Institute G. M. ofde Chemical, Oliveira Biological,1, Roberto Radiological B. Sousa and1, Paulo Nuclear A. Defense de M. Cabral (IDQBRN),1, Kamil Brazilian Kuˇca Army3,* and TanosTechnological C. C. França Center3,4,* (CTEx), Avenida das Américas 28705, Rio de Janeiro 23020-470, Brazil; [email protected] (M.C.B.); [email protected] (V.G.M.d.O.); [email protected] 1 Institute of Chemical, Biological, Radiological and Nuclear Defense (IDQBRN), Brazilian Army (R.B.S.); [email protected] (P.A.d.M.C.) Technological Center (CTEx), Avenida das Américas 28705, Rio de Janeiro 23020-470, Brazil; 2 [email protected] Mors Institute of Research (M.C.B.); on Natural [email protected] Products (IPPN), Federal (V.G.M.d.O.); University of Rio de Janeiro (UFRJ), CCS,[email protected] Bloco H, Rio de Janeiro (R.B.S.); 21941-902, [email protected] Brazil (P.A.d.M.C.) 32 DepartmentWalter Mors of Institute Chemistry, of Research Faculty of on Science, Natural Un Productsiversity (IPPN), -

Department of Homeland Security (Dhs) Appendix a Chemicals of Interest (Coi)
DEPARTMENT OF HOMELAND SECURITY (DHS) APPENDIX A CHEMICALS OF INTEREST (COI) Acetaldehyde Bromine trifluoride Acetone Cyanohydrin, stabilized Bromothrifluorethylene Acetyl bromide 1,3-Butadiene Acetyl chloride Butane Acetyl iodide Butene Acetylene 1-Butene Acrolein 2-Butene Acrylonitrile 2-Butene-cis Acrylyl chloride 2-Butene-trans Allyl alcohol Butyltrichlorosilane Allylamine Calcium hydrosulfite Allyltrichlorosilane, stabilized Calcium phosphide Aluminum (powder) Carbon disulfide Aluminum Bromide, anhydrous Carbon oxysulfide Aluminum Chloride, anhydrous Carbonyl fluoride Aluminum phosphide Carbonyl sulfide Ammonia (anhydrous) Chlorine Ammonia (conc 20% or greater) Chlorine dioxide Ammonium nitrate, note #1 Chlorine monoxide Ammonium nitrate, note #2 Chlorine pentafluoride Ammonium perchlorate Chlorine trifluoride Ammonium picrate Chloroacetyl chloride Amyltrichlorosilane 2-Chloroethylchloro-methylsulfide Antimony pentafluoride Chloroform Arsenic trichloride Chloromethyl ether Arsine Chloromethyl methyl ether Barium azide 1-Chloropropylene 1,4-Bis(2-chloroethylthio)-n-butane 2-Chloropropylene Bis(2-chloroethylthio) methane Chlorosarin Bis(2-chloroethylthiomethyl)ether Chlorosoman 1,5-Bis(2-chloroethylthio)-n-pentane Chlorosulfonic acid 1,3-Bis(2-chloroethylthio)-n-propane Chromium oxychloride Boron tribromide Crotonaldehyde Boron trichloride Crotonaldehyde, (E)- Boron trifluoride Cyanogen Boron trifluoride compound with methyl Cyanogen chloride ether (1:1) Cyclohexylamine Bromine Cyclohexyltrichlorosilane Bromine chloride Cyclopropane -

Export Control Handbook for Chemicals
Export Control Handbook for Chemicals -Dual-use control list -Common Military List -Explosives precursors -Syria restrictive list -Psychotropics and narcotics precursors ARNES-NOVAU, X 2019 EUR 29879 This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Xavier Arnés-Novau Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-785421 Filippo Sevini Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-786793 EU Science Hub https://ec.europa.eu/jrc JRC 117839 EUR 29879 Print ISBN 978-92-76-11971-5 ISSN 1018-5593 doi:10.2760/844026 PDF ISBN 978-92-76-11970-8 ISSN 1831-9424 doi:10.2760/339232 Luxembourg: Publications Office of the European Union, 2019 © European Atomic Energy Community, 2019 The reuse policy of the European Commission is implemented by Commission Decision 2011/833/EU of 12 December 2011 on the reuse of Commission documents (OJ L 330, 14.12.2011, p. 39). Reuse is authorised, provided the source of the document is acknowledged and its original meaning or message is not distorted. The European Commission shall not be liable for any consequence stemming from the reuse. -

CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011
CSAT Top-Screen Questions January 2009 Version 2.8 CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011 Change Log .........................................................................................................3 CVI Authorizing Statements...............................................................................4 General ................................................................................................................6 Facility Description.................................................................................................................... 7 Facility Regulatory Mandates ................................................................................................... 7 EPA RMP Facility Identifier....................................................................................................... 9 Refinery Capacity....................................................................................................................... 9 Refinery Market Share ............................................................................................................. 10 Airport Fuels Supplier ............................................................................................................. 11 Military Installation Supplier................................................................................................... 11 Liquefied Natural Gas (LNG) Capacity................................................................................... 12 Liquefied Natural Gas Exclusion -

Chemical Weapons Technology Section 4—Chemical Weapons Technology
SECTION IV CHEMICAL WEAPONS TECHNOLOGY SECTION 4—CHEMICAL WEAPONS TECHNOLOGY Scope Highlights 4.1 Chemical Material Production ........................................................II-4-8 4.2 Dissemination, Dispersion, and Weapons Testing ..........................II-4-22 • Chemical weapons (CW) are relatively inexpensive to produce. 4.3 Detection, Warning, and Identification...........................................II-4-27 • CW can affect opposing forces without damaging infrastructure. 4.4 Chemical Defense Systems ............................................................II-4-34 • CW can be psychologically devastating. • Blister agents create casualties requiring attention and inhibiting BACKGROUND force efficiency. • Defensive measures can be taken to negate the effect of CW. Chemical weapons are defined as weapons using the toxic properties of chemi- • Donning of protective gear reduces combat efficiency of troops. cal substances rather than their explosive properties to produce physical or physiologi- • Key to employment is dissemination and dispersion of agents. cal effects on an enemy. Although instances of what might be styled as chemical weapons date to antiquity, much of the lore of chemical weapons as viewed today has • CW are highly susceptible to environmental effects (temperature, its origins in World War I. During that conflict “gas” (actually an aerosol or vapor) winds). was used effectively on numerous occasions by both sides to alter the outcome of • Offensive use of CW complicates command and control and battles. A significant number of battlefield casualties were sustained. The Geneva logistics problems. Protocol, prohibiting use of chemical weapons in warfare, was signed in 1925. Sev- eral nations, the United States included, signed with a reservation forswearing only the first use of the weapons and reserved the right to retaliate in kind if chemical weapons were used against them. -

Synthesis of Nerve Agent- and Pesticide-Protein Bioadducts As Reference Materials for Retrospective Verification of Exposure
University of Fribourg / Faculty of Science and Medicine / Department of Chemistry Synthesis of Nerve Agent- and Pesticide-Protein Bioadducts as Reference Materials for Retrospective Verification of Exposure Andreas Bielmann Cholinesterase inhibiting organophosphorus compounds are highly toxic and are used as pesticides or nerve agents. As most chemical warfare agents, nerve agents are prohibited by the Chemical Weapons Convention. To enforce the Convention, the Organisation for the Prohibition of Chemical Weapons maintains a network of laboratories, which are designated to analyse samples after alleged incidents involving chemical warfare agents. The gold standard for the retrospective verification of exposure to nerve agents in biomedical samples is the LC-MS analysis of a specific nonapeptide with the sequence FGES*AGAAS obtained after pepsin digestion of butyrylcholinesterase. Organophosphorus inhibitors will leave a specific marker on the serine indicated by *. Reference materials for this analysis are often produced by spiking of blood with agents. However, this method only gives reference peptides of inadequate purity. For the development of analytical methods and for quantitative analysis highly pure synthetic peptides are required. In this work, the synthesis of phosphylated peptides via a building block approach was investigated. Synthesis protocols were established and the limitations of the method were investigated. Phosphylated amino acid precursors and the resulting peptides have some intrinsic labilities, therefore an optimized SPPS protocol was developed to overcome them. Using this protocol, two pathways to synthesize the aged-nonapeptide adduct were investigated and compared. Further, the bioadducts of the nerve agents sarin, cyclosarin, VX, CVX and RVX, as well as the methyl methylphosphono- and isopropyl isopropylphosphono nonapeptides were synthesized to determine the scope of the method. -

Central Intelligence Agency (CIA) Document: the Biological and Chemical Warfare Threat, January 1997
Description of document: Central Intelligence Agency (CIA) document: The Biological and Chemical Warfare Threat, January 1997 Requested date: 01-July-2015 Released date: 02-December-2015 Posted date: 04-January-2016 Source of document: Freedom of Information Act Request Information and Privacy Coordinator Central Intelligence Agency Washington, D.C. 20505 Fax: 703-613-3007 Filing a FOIA Records Request Online The governmentattic.org web site (“the site”) is noncommercial and free to the public. The site and materials made available on the site, such as this file, are for reference only. The governmentattic.org web site and its principals have made every effort to make this information as complete and as accurate as possible, however, there may be mistakes and omissions, both typographical and in content. The governmentattic.org web site and its principals shall have neither liability nor responsibility to any person or entity with respect to any loss or damage caused, or alleged to have been caused, directly or indirectly, by the information provided on the governmentattic.org web site or in this file. The public records published on the site were obtained from government agencies using proper legal channels. Each document is identified as to the source. Any concerns about the contents of the site should be directed to the agency originating the document in question. GovernmentAttic.org is not responsible for the contents of documents published on the website. Central Intelligence Agency Washington,• D.C. 20505 2 December 2015 Reference: F-2015-02095 This is a final response to your 1July2015 Freedom of Information Act (FOIA) request for "a copy of the following six CIA documents: 1. -

Chemicals Requiring EHS Pre-Approval
Chemicals Requiring EHS Approval Before Purchasing Chemical Name CAS # 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 1H-Tetrazole 288-94-8 2-chloroethyl ethylsulfide 693-07-2 2-Chloroethylchloro-methylsulfide 2625-76-5 2-Cyanoethyl diisopropylchlorophosphoramidite 89992-70-1 2-Ethoxyethanol 110-80-5 2-Ethoxyethylacetate 111-15-9 2-Methoxyethanol 109-86-4 2-Methoxyethylacetate 110-49-6 5-Nitrobenzotriazol 2338-12-7 Acetone cyanohydrin, stabilized 75-86-5 Acrolein 107-02-8 Acrylamide (powder) 79-06-1 Allylamine 107-11-9 Aluminum (powder) 7429-90-5 Aluminum phosphide 20859-73-8 Ammonia 7664-41-7 Ammonium nitrate 6484-52-2 Ammonium perchlorate 7790-98-9 Ammonium picrate 131-74-8 Arsenic 7440-38-2 Arsenic trichloride 7784-34-1 Arsenic trioxide 1327-53-3 Arsine 7784-42-1 Barium azide 18810-58-7 Beryllium 7440-41-7 Bis(2-chloroethylthio)methane 63869-13-6 Bis(2-chloroethylthiomethyl)ether 63918-90-1 Boron tribromide 10294-33-4 Boron trichloride 10294-34-5 Boron trifluoride 7637-07-2 Bromine 7726-95-6 Bromine chloride 13863-41-7 Bromine pentafluoride 7789-30-2 Bromine trifluoride 7787-71-5 Cadmium 7440-43-9 Calcium phosphide 1305-99-3 Carbon monoxide 630-08-0 Carbonyl fluoride 353-50-4 Carbonyl sulfide 463-58-1 Chlorine 7782-50-5 Chlorine dioxide 10049-04-4 Chlorine pentafluoride 13637-63-3 Chlorine trifluoride 7790-91-2 Chemicals Requiring EHS Approval Before Purchasing Chloroacetyl chloride 79-04-9 Chlorosarin 1445-76-7 Chlorosoman 7040-57-5 Chlorosulfonic