Endogenous and Exogenous Ligands of Aryl Hydrocarbon Receptor: Current State of Art
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

Open Thesis Master Document V5.0.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Science IDENTIFICATION OF ENDOGENOUS MODULATORS FOR THE ARYL HYDROCARBON RECEPTOR A Thesis in Genetics by Christopher R. Chiaro © 2007 Christopher R. Chiaro Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy December, 2007 The thesis of Christopher R. Chiaro was reviewed and approved* by the following: Gary H. Perdew John T. and Paige S. Smith Professor in Agricultural Sciences Thesis Advisor Chair of Committee C. Channa Reddy Distinguished Professor of Veterinary Science A. Daniel Jones Senior Scientist Department of Chemistry John P. Vanden Heuvel Professor of Veterinary Science Richard Ordway Associate Professor of Biology Chair of Genetics Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor capable of being regulated by a structurally diverse array of chemicals ranging from environmental carcinogens to dietary metabolites. A member of the basic helix-loop- helix/ Per-Arnt-Sim (bHLH-PAS) super-family of DNA binding regulatory proteins, the AhR is an important developmental regulator that can be detected in nearly all mammalian tissues. Prior to ligand activation, the AhR resides in the cytosol as part of an inactive oligomeric protein complex comprised of the AhR ligand-binding subunit, a dimer of the 90 kDa heat shock protein, and a single molecule each of the immunophilin like X-associated protein 2 (XAP2) and p23 proteins. Functioning as chemosensor, the AhR responds to both endobiotic and xenobiotic derived chemical ligands by ultimately directing the expression of metabolically important target genes. -

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Independent Actions on Cyclin-Dependent Kinases and Aryl Hydrocarbon Receptor Mediate the Antiproliferative Effects of Indirubins
Oncogene (2004) 23, 4400–4412 & 2004 Nature Publishing Group All rights reserved 0950-9232/04 $30.00 www.nature.com/onc Independent actions on cyclin-dependent kinases and aryl hydrocarbon receptor mediate the antiproliferative effects of indirubins Marie Knockaert1, Marc Blondel1, Ste´ phane Bach1, Maryse Leost1, Cem Elbi2, Gordon L Hager2, Scott RNagy 3, Dalho Han3, Michael Denison3, Martine Ffrench4, Xiaozhou P Ryan5, Prokopios Magiatis6, Panos Polychronopoulos6, Paul Greengard5, Leandros Skaltsounis6 and Laurent Meijer*,1,5 1C.N.R.S., Cell Cycle Group & UPS-2682, Station Biologique, BP 74, 29682 ROSCOFF cedex, Bretagne, France; 2Laboratory of Receptor Biology & Gene Expression, Bldg 41, Room B602, National Cancer Institute, NIH, Bethesda, MD 20892-5055, USA; 3Department of Environmental Toxicology, Meyer Hall, One Shields Avenue, University of California, Davis, CA 95616-8588, USA; 4Universite´ Claude Bernard, Laboratoire de Cytologie Analytique et Cytoge´ne´tique Mole´culaire, 8 Avenue Rockefeller, 69373 Lyon Cedex 08, France; 5Laboratory of Molecular & Cellular Neuroscience, The Rockefeller University, 1230 York Avenue, New York, NY 10021, USA; 6Division of Pharmacognosy and Natural Products Chemistry, Department of Pharmacy, University of Athens, Panepistimiopolis Zografou, GR-15771 Athens, Greece Indirubin, a bis-indole obtained from various natural Introduction sources, is responsible for the reported antileukemia activity of a Chinese Medicinal recipe, Danggui Longhui The bis-indole indirubin has been identified as the main Wan. However, its molecular mechanism of action is still active ingredient of a traditional Chinese medicinal not well understood. In addition to inhibition of cyclin- recipe, Danggui Longhui Wan, used to treat various dependent kinases and glycogen synthase kinase-3, diseases including chronic myelocytic leukemia (reviews indirubins have been reported to activate the aryl in Tang and Eisenbrand, 1992; Xiao et al., 2002). -

Detection, Occurrence and Fate of Indirubin in Municipal Sewage
Subscriber access provided by BEIJING UNIV Article Detection, Occurrence and Fate of Indirubin in Municipal Sewage Treatment Plants Jianying Hu, Hong Chang, Lezheng Wang, Shimin Wu, Bin Shao, Jun Zhou, and Ying Zhao Environ. Sci. Technol., 2008, 42 (22), 8339-8344• DOI: 10.1021/es801038y • Publication Date (Web): 11 October 2008 Downloaded from http://pubs.acs.org on March 12, 2009 More About This Article Additional resources and features associated with this article are available within the HTML version: • Supporting Information • Access to high resolution figures • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article Environmental Science & Technology is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 Environ. Sci. Technol. 2008, 42, 8339–8344 to cause liver damage, growth retardation, certain types of Detection, Occurrence and Fate of cancer, birth defects, depression of immunological function, Indirubin in Municipal Sewage and reproductive problems in several species by binding to activating aryl hydrocarbon receptor (AhR) (3-6). These AhR Treatment Plants agonists have been identified in almost every component of the global ecosystem including fish, wildlife, human adipose tissue, milk, and serum (7). JIANYING HU,*,† HONG CHANG,† LEZHENG WANG,† SHIMIN WU,† Besides these chemicals, a wide variety of naturally BIN SHAO,‡ JUN ZHOU,§ AND occurring chemicals such as tetrapyroles, indoles, and YING ZHAO§ arachidonic acid metabolites have been also reported to be College of Urban and Environmental Science, Peking AhR agonists (8, 9). Some of these chemicals were reported University, Beijing 100871, China, Institute of Nutrition and to interact with AhR and produce activation or inhibition of Food Hygiene, Beijing Center for Disease Prevention and signal transduction. -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

Bilirubin and Beyond: a Review of Lipid Status in Gilbert's Syndrome and Its Relevance to Cardiovascular Disease Protection
Bilirubin and beyond: A review of lipid status in Gilbert's syndrome and its relevance to cardiovascular disease protection Author Bulmer, AC, Verkade, HJ, Wagner, K-H Published 2013 Journal Title Progress in Lipid Research DOI https://doi.org/10.1016/j.plipres.2012.11.001 Copyright Statement © 2013 Elsevier. This is the author-manuscript version of this paper. Reproduced in accordance with the copyright policy of the publisher. Please refer to the journal's website for access to the definitive, published version. Downloaded from http://hdl.handle.net/10072/54228 Griffith Research Online https://research-repository.griffith.edu.au Bilirubin and beyond: a review of lipid status in Gilbert’s syndrome and its relevance to cardiovascular disease protection Bulmer, A.C.1*, Verkade, H.J.2, Wagner, K-H3* 1Heart Foundation Research Centre, Griffith Health Institute, Griffith University, Gold Coast, Australia. 2Pediatric Gastroenterology – Hepatology, Beatrix Children’s Hospital, University Medical Center Groningen, University of Groningen, The Netherlands. 3Emerging Field Oxidative Stress and DNA Stability, Department of Nutritional Sciences, University of Vienna, Vienna, Austria. *Corresponding Authors Prof Karl-Heinz Wagner Department of Nutritional Sciences Althanstrasse 14, 1090, Vienna, Austria Ph: +43 1 427754930 Fax: +43 1 42779549 Dr Andrew Bulmer Heart Foundation Research Centre Griffith Health Institute Griffith University Parklands Avenue, Southport, Southport, 4222, Australia Ph: +61 7 55528215 Fax: +61 7 5552 8908 1 Abstract Gilbert’s syndrome (GS) is characterized by a benign, mildly elevated bilirubin concentration in the blood. Recent reports show clear protection from cardiovascular disease in this population. Protection of lipids, proteins and other macromolecules from oxidation by bilirubin represents the most commonly accepted mechanism contributing to protection in this group. -
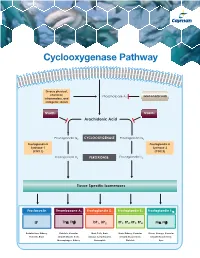
Cyclooxygenase Pathway
Cyclooxygenase Pathway Diverse physical, chemical, Phospholipase A Glucocorticoids inflammatory, and 2 mitogenic stimuli NSAIDs NSAIDs Arachidonic Acid Prostaglandin G2 CYCLOOXYGENASE Prostaglandin G2 Prostaglandin H Prostaglandin H Synthase-1 Synthase-2 (COX 1) (COX 2) Prostaglandin H2 PEROXIDASE Prostaglandin H2 Tissue Specific Isomerases Prostacyclin Thromboxane A2 Prostaglandin D2 Prostaglandin E2 Prostaglandin F2α IP TPα, TPβ DP1, DP2 EP1, EP2, EP3, EP4 FPα, FPβ Endothelium, Kidney, Platelets, Vascular Mast Cells, Brain, Brain, Kidney, Vascular Uterus, Airways, Vascular Platelets, Brain Smooth Muscle Cells, Airways, Lymphocytes, Smooth Muscle Cells, Smooth Muscle Cells, Macrophages, Kidney Eosinophils Platelets Eyes Prostacyclin Item No. Product Features Prostacyclin (Prostaglandin I2; PGI2) is formed from arachidonic acid primarily in the vascular endothelium and renal cortex by sequential 515211 6-keto • Sample Types: Culture Medium | Plasma Prostaglandin • Measure 6-keto PGF levels down to 6 pg/ml activities of COX and prostacyclin synthase. PGI2 is non-enzymatically 1α F ELISA Kit • Incubation : 18 hours | Development: 90-120 minutes | hydrated to 6-keto PGF1α (t½ = 2-3 minutes), and then quickly converted 1α Read: Colorimetric at 405-420 nm to the major metabolite, 2,3-dinor-6-keto PGF1α (t½= 30 minutes). Prostacyclin was once thought to be a circulating hormone that regulated • Assay 24 samples in triplicate or 36 samples in duplicate platelet-vasculature interactions, but the rate of secretion into circulation • NOTE: A portion of urinary 6-keto PGF1α is of renal origin coupled with the short half-life indicate that prostacyclin functions • NOTE : It has been found that normal plasma levels of 6-keto PGF may be low locally. -

Dioxin) Receptor
TOXICOLOGICAL SCIENCES 124(1), 1–22 (2011) doi:10.1093/toxsci/kfr218 Advance Access publication September 9, 2011 Exactly the Same but Different: Promiscuity and Diversity in the Molecular Mechanisms of Action of the Aryl Hydrocarbon (Dioxin) Receptor Michael S. Denison,*,1 Anatoly A. Soshilov,* Guochun He,* Danica E. DeGroot,* and Bin Zhao† Downloaded from *Department of Environmental Toxicology, University of California, Davis, California 95616; and †Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, China 1To whom correspondence should be addressed at Department of Environmental Toxicology, University of California, Meyer Hall, One Shields Avenue, Davis, CA 95616-8588. Fax: (530) 752-3394. E-mail: [email protected]. http://toxsci.oxfordjournals.org/ Received June 6, 2011; accepted August 12, 2011 toxic effects in a wide range of species and tissues (Beischlag The Ah receptor (AhR) is a ligand-dependent transcription et al., 2008; Furness and Whelan, 2009; Hankinson, 2005; factor that mediates a wide range of biological and toxicological Humblet et al., 2008; Marinkovic´ et al., 2010; Poland and effects that result from exposure to a structurally diverse variety Knutson, 1982; Safe, 1990; White and Birnbaum, 2009). The of synthetic and naturally occurring chemicals. Although the overall mechanism of action of the AhR has been extensively best-characterized high-affinity ligands for the AhR include at University of California, Davis - Library on May 4, 2012 studied and involves a classical nuclear receptor mechanism of a wide variety of ubiquitous hydrophobic environmental action (i.e., ligand-dependent nuclear localization, protein hetero- contaminants such as the halogenated aromatic hydrocarbons dimerization, binding of liganded receptor as a protein complex to (HAHs) and nonhalogenated polycyclic aromatic hydrocarbons its specific DNA recognition sequence and activation of gene (PAHs). -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Pyrene in the Amphibian Xenopus Laevis
DEVELOPMENTAL SENSITIVITY AND IMMUNOTOXICITY OF BENZO[A]PYRENE IN THE AMPHIBIAN XENOPUS LAEVIS Melanie J. Gallant A Thesis Submitted to the College of Graduate and Postdoctoral Studies In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy In the Toxicology Graduate Program University of Saskatchewan Saskatoon, Saskatchewan, Canada © Copyright Melanie Gallant, 2019. All rights reserved PERMISSION TO USE In presenting this thesis in partial fulfillment of the requirements for a postgraduate degree from the University of Saskatchewan, I agree that the Libraries of the University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purpose may be granted by the professors who supervised my thesis work or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that any copying or publication of use of this thesis or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due recognition shall be given to me and the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or parts should be addressed to: Chair of the Toxicology Graduate Program Toxicology Centre, University of Saskatchewan 44 Campus Drive Saskatoon, Saskatchewan S7N 5B3 OR Dean College of Graduate and Postdoctoral Studies University of Saskatchewan 116 Thorvaldson Building, 110 Science Place Saskatoon, Saskatchewan S7N 5C9 Canada i ABSTRACT The immune system is increasingly recognized as a vulnerable and sensitive target of contaminant exposure. -

THE EFFECTS of INDIRUBIN DERIVITIVES on GENE EXPRESSION and CELLULAR FUNCTIONS in the MURINE MACROPHAGE Abigail Babcock Clemson University, [email protected]
Clemson University TigerPrints All Dissertations Dissertations 8-2008 THE EFFECTS OF INDIRUBIN DERIVITIVES ON GENE EXPRESSION AND CELLULAR FUNCTIONS IN THE MURINE MACROPHAGE Abigail Babcock Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations Part of the Biology Commons Recommended Citation Babcock, Abigail, "THE EFFECTS OF INDIRUBIN DERIVITIVES ON GENE EXPRESSION AND CELLULAR FUNCTIONS IN THE MURINE MACROPHAGE" (2008). All Dissertations. 277. https://tigerprints.clemson.edu/all_dissertations/277 This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations by an authorized administrator of TigerPrints. For more information, please contact [email protected]. THE EFFECTS OF INDIRUBIN DERIVATIVES ON GENE EXPRESSION AND CELLULAR FUNCTIONS IN THE MURINE CELL LINE, RAW 264.7 A Dissertation Presented to the Graduate School of Clemson University In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Biological Sciences by Abigail Sue Babcock August 2008 Accepted by: Dr. Charles D. Rice, Committee Chair Dr. Lyndon L. Larcom Dr. Thomas R. Scott Dr. Alfred P. Wheeler ABSTRACT Indirubin is the active ingredient in the ancient herbal remedy, Danggui Longhui Wan, which is used as an anti-leukemic, anti-inflammatory, and detoxification treatment. Indirubin-3’-monoxime (IO) is the most widely studied of the indirubin derivatives. Most have been shown to inhibit cyclin-dependent kinases and glycogen synthase kinases, triggering cell cycle arrest and apoptosis with different levels of activity depending on the particular structure. These kinases inhibited by indirubin(s) play key roles in cellular proliferation and immune functions, therefore synthetic indirubins may have significant applications as therapeutic agents for cancer, inflammation, and neurological diseases.