Full Text of Thesis (PDF, 4.091Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstruction of the Global Neural Crest Gene Regulatory Network in Vivo
Reconstruction of the global neural crest gene regulatory network in vivo Ruth M Williams1, Ivan Candido-Ferreira1, Emmanouela Repapi2, Daria Gavriouchkina1,4, Upeka Senanayake1, Jelena Telenius2,3, Stephen Taylor2, Jim Hughes2,3, and Tatjana Sauka-Spengler1,∗ Supplemental Material ∗Lead and corresponding author: Tatjana Sauka-Spengler ([email protected]) 1University of Oxford, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, Oxford, OX3 9DS, UK 2University of Oxford, MRC Centre for Computational Biology, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 3University of Oxford, MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 4Present Address: Okinawa Institute of Science and Technology, Molecular Genetics Unit, Onna, 904-0495, Japan A 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R1_5-6ss) log2(R1_8-10ss) log2(R1_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.92 0 r=0.99 0 r=0.96 0 r=0.99 0 r=0.96 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R2_non-NC) log2(R2_5-6ss) log2(R3_5-6ss) log2(R2_8-10ss) log2(R3_8-10ss) 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R2_5-6ss) log2(R1_8-10ss) log2(R2_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.94 0 r=0.96 0 r=0.95 0 r=0.96 0 r=0.95 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R3_non-NC) log2(R4_5-6ss) log2(R3_5-6ss) log2(R4_8-10ss) log2(R3_8-10ss) -

Gene PMID WBS Locus ABR 26603386 AASDH 26603386
Supplementary material J Med Genet Gene PMID WBS Locus ABR 26603386 AASDH 26603386 ABCA1 21304579 ABCA13 26603386 ABCA3 25501393 ABCA7 25501393 ABCC1 25501393 ABCC3 25501393 ABCG1 25501393 ABHD10 21304579 ABHD11 25501393 yes ABHD2 25501393 ABHD5 21304579 ABLIM1 21304579;26603386 ACOT12 25501393 ACSF2,CHAD 26603386 ACSL4 21304579 ACSM3 26603386 ACTA2 25501393 ACTN1 26603386 ACTN3 26603386;25501393;25501393 ACTN4 21304579 ACTR1B 21304579 ACVR2A 21304579 ACY3 19897463 ACYP1 21304579 ADA 25501393 ADAM12 21304579 ADAM19 25501393 ADAM32 26603386 ADAMTS1 25501393 ADAMTS10 25501393 ADAMTS12 26603386 ADAMTS17 26603386 ADAMTS6 21304579 ADAMTS7 25501393 ADAMTSL1 21304579 ADAMTSL4 25501393 ADAMTSL5 25501393 ADCY3 25501393 ADK 21304579 ADRBK2 25501393 AEBP1 25501393 AES 25501393 AFAP1,LOC84740 26603386 AFAP1L2 26603386 AFG3L1 21304579 AGAP1 26603386 AGAP9 21304579 Codina-Sola M, et al. J Med Genet 2019; 56:801–808. doi: 10.1136/jmedgenet-2019-106080 Supplementary material J Med Genet AGBL5 21304579 AGPAT3 19897463;25501393 AGRN 25501393 AGXT2L2 25501393 AHCY 25501393 AHDC1 26603386 AHNAK 26603386 AHRR 26603386 AIDA 25501393 AIFM2 21304579 AIG1 21304579 AIP 21304579 AK5 21304579 AKAP1 25501393 AKAP6 21304579 AKNA 21304579 AKR1E2 26603386 AKR7A2 21304579 AKR7A3 26603386 AKR7L 26603386 AKT3 21304579 ALDH18A1 25501393;25501393 ALDH1A3 21304579 ALDH1B1 21304579 ALDH6A1 21304579 ALDOC 21304579 ALG10B 26603386 ALG13 21304579 ALKBH7 25501393 ALPK2 21304579 AMPH 21304579 ANG 21304579 ANGPTL2,RALGPS1 26603386 ANGPTL6 26603386 ANK2 21304579 ANKMY1 26603386 ANKMY2 -
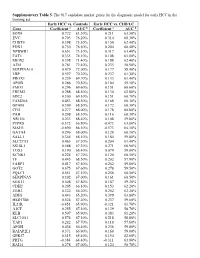
Supplementary Table 5. the 917 Candidate Marker Genes for the Diagnostic Model for Early HCC in the Training Set
Supplementary Table 5. The 917 candidate marker genes for the diagnostic model for early HCC in the training set. Early HCC vs. Controls Early HCC vs. CHB/LC Gene Coefficient a AUC b Coefficient a AUC b SOX9 0.722 81.30% 0.211 63.50% EVC 0.703 76.20% 0.314 68.30% CHST9 0.398 75.50% 0.150 62.40% PDX1 0.730 76.50% 0.204 60.40% NPBWR1 0.651 73.10% 0.317 63.40% FAT1 0.335 74.10% 0.108 61.00% MEIS2 0.398 71.40% 0.188 62.40% A2M 0.761 72.40% 0.235 58.90% SERPINA10 0.479 72.00% 0.177 58.40% LBP 0.597 70.20% 0.237 61.30% PROX1 0.239 69.70% 0.133 61.40% APOB 0.286 70.50% 0.104 59.10% FMO3 0.296 69.60% 0.151 60.60% FREM2 0.288 68.50% 0.130 62.80% SDC2 0.300 69.10% 0.151 60.70% FAM20A 0.453 68.50% 0.168 60.30% GPAM 0.309 68.50% 0.172 60.30% CFH 0.277 68.00% 0.178 60.80% PAH 0.208 68.30% 0.116 60.30% NR1H4 0.233 68.40% 0.108 59.80% PTPRS -0.572 66.80% -0.473 63.00% SIAH3 -0.690 66.10% -0.573 64.10% GATA4 0.296 68.00% 0.128 60.10% SALL1 0.344 68.10% 0.184 59.80% SLC27A5 0.463 67.30% 0.204 61.40% SS18L1 0.588 67.30% 0.271 60.90% TOX3 0.190 68.40% 0.079 59.00% KCNK1 0.224 67.70% 0.120 60.10% TF 0.445 68.50% 0.202 57.90% FARP1 0.417 67.50% 0.252 59.80% GOT2 0.675 67.60% 0.278 59.50% PQLC1 0.651 67.10% 0.258 60.50% SERPINA5 0.302 67.00% 0.161 60.50% SOX13 0.508 67.80% 0.187 59.30% CDH2 0.205 66.10% 0.153 62.20% ITIH2 0.322 66.20% 0.252 62.20% ADIG 0.443 65.20% 0.399 63.80% HSD17B6 0.524 67.20% 0.237 59.60% IL21R -0.451 65.90% -0.321 61.70% A1CF 0.255 67.10% 0.139 58.70% KLB 0.507 65.90% 0.383 61.20% SLC10A1 0.574 67.10% 0.218 58.80% YAP1 0.282 67.70% 0.118 -

Prioritization of Metabolic Genes As Novel Therapeutic Targets in Estrogen-Receptor Negative Breast Tumors Using Multi-Omics Data and Text Mining
www.oncotarget.com Oncotarget, 2019, Vol. 10, (No. 39), pp: 3894-3909 Research Paper Prioritization of metabolic genes as novel therapeutic targets in estrogen-receptor negative breast tumors using multi-omics data and text mining Dinesh Kumar Barupal1,*, Bei Gao1,*, Jan Budczies2, Brett S. Phinney4, Bertrand Perroud4, Carsten Denkert2,3 and Oliver Fiehn1 1West Coast Metabolomics Center, University of California, Davis, CA, USA 2Institute of Pathology, Charité University Hospital, Berlin, Germany 3German Institute of Pathology, Philipps-University Marburg, Marburg, Germany 4UC Davis Genome Center, University of California, Davis, CA, USA *Co-first authors and contributed equally to this work Correspondence to: Oliver Fiehn, email: [email protected] Keywords: set-enrichment; ChemRICH; multi-omics; metabolic networks; candidate gene prioritization Received: March 12, 2019 Accepted: May 13, 2019 Published: June 11, 2019 Copyright: Barupal et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Estrogen-receptor negative (ERneg) breast cancer is an aggressive breast cancer subtype in the need for new therapeutic options. We have analyzed metabolomics, proteomics and transcriptomics data for a cohort of 276 breast tumors (MetaCancer study) and nine public transcriptomics datasets using univariate statistics, meta- analysis, Reactome pathway analysis, biochemical network mapping and text mining of metabolic genes. In the MetaCancer cohort, a total of 29% metabolites, 21% proteins and 33% transcripts were significantly different (raw p <0.05) between ERneg and ERpos breast tumors. -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

Clinical Characterization of Chromosome 5Q21.1–21.3 Microduplication: a Case Report
Open Medicine 2020; 15: 1123–1127 Case Report Shuang Chen, Yang Yu, Han Zhang, Leilei Li, Yuting Jiang, Ruizhi Liu, Hongguo Zhang* Clinical characterization of chromosome 5q21.1–21.3 microduplication: A case report https://doi.org/10.1515/med-2020-0199 Keywords: chromosome 5, prenatal diagnosis, microdu- received May 20, 2020; accepted September 28, 2020 plication, genetic counseling Abstract: Chromosomal microdeletions and microdupli- cations likely represent the main genetic etiologies for children with developmental delay or intellectual dis- ability. Through prenatal chromosomal microarray ana- 1 Introduction lysis, some microdeletions or microduplications can be detected before birth to avoid unnecessary abortions or Chromosomal microdeletions,microduplications,and birth defects. Although some microdeletions or microdu- unbalanced rearrangements represent the main genetic plications of chromosome 5 have been reported, nu- etiological factors for children with developmental delay [ ] - merous microduplications remain undescribed. We de- or intellectual disability 1 . Currently, chromosomal mi ( ) fi - - scribe herein a case of a 30-year-old woman carrying a croarray analysis CMA is considered a rst tier diag [ ] - fetus with a chromosome 5q21.1–q21.3 microduplication. nostic tool for these children 2 . Through prenatal diag Because noninvasive prenatal testing indicated a fetal nosis of CMA, some microdeletions or microduplications - chromosome 5 abnormality, the patient underwent am- can be detected before birth to avoid unnecessary abor [ ] niocentesis at 22 weeks 4 days of gestation. Karyotyping tions or birth defects 3 . The clinical features of some and chromosomal microarray analysis were performed on chromosome 5 microduplications have been described [ – ] [ ] amniotic fluid cells. Fetal behavioral and structural ab- previously 4 8 . -

Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks
bioRxiv preprint doi: https://doi.org/10.1101/219667; this version posted November 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Article Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks Weiguang Mao 1,2, Dennis Kostka 1,2,3 and Maria Chikina 1,2* 1 Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh 2 Joint Carnegie Mellon - University of Pittsburgh Ph.D. Program in Computational Biology 3 Department of Developmental Biology, School of Medicine, University of Pittsburgh * Correspondence: [email protected] Academic Editor: name Version November 9, 2017 submitted to Cells 1 Abstract: Background: Gene regulatory sequences play critical roles in ensuring tightly controlled 2 RNA expression patterns that are essential in a large variety of biological processes. Specifically, 3 enhancer sequences drive expression of their target genes, and the availability of genome-wide 4 maps of enhancer-promoter interactions has opened up the possibility to use machine learning 5 approaches to extract and interpret features that define these interactions in different biological 6 contexts. Methods: Inspired by machine translation models we develop an attention-based neural 7 network model, EPIANN, to predict enhancer-promoter interactions based on DNA sequences. Codes 8 and data are available at https://github.com/wgmao/EPIANN. Results: Our approach accurately 9 predicts enhancer-promoter interactions across six cell lines. In addition, our method generates 10 pairwise attention scores at the sequence level, which specify how short regions in the enhancer and 11 promoter pair-up to drive the interaction prediction. -

Biological Pathways, Candidate Genes, and Molecular Markers Associated with Quality-Of-Life Domains: an Update
Qual Life Res (2014) 23:1997–2013 DOI 10.1007/s11136-014-0656-1 REVIEW Biological pathways, candidate genes, and molecular markers associated with quality-of-life domains: an update Mirjam A. G. Sprangers • Melissa S. Y. Thong • Meike Bartels • Andrea Barsevick • Juan Ordon˜ana • Qiuling Shi • Xin Shelley Wang • Pa˚l Klepstad • Eddy A. Wierenga • Jasvinder A. Singh • Jeff A. Sloan Accepted: 19 February 2014 / Published online: 7 March 2014 Ó Springer International Publishing Switzerland 2014 Abstract (depressed mood) and positive (well-being/happiness) Background There is compelling evidence of a genetic emotional functioning, social functioning, and overall foundation of patient-reported quality of life (QOL). Given QOL. the rapid development of substantial scientific advances in Methods We followed a purposeful search algorithm of this area of research, the current paper updates and extends existing literature to capture empirical papers investigating reviews published in 2010. the relationship between biological pathways and molecu- Objectives The objective was to provide an updated lar markers and the identified QOL domains. overview of the biological pathways, candidate genes, and Results Multiple major pathways are involved in each molecular markers involved in fatigue, pain, negative QOL domain. The inflammatory pathway has the strongest evidence as a controlling mechanism underlying fatigue. Inflammation and neurotransmission are key processes On behalf of the GeneQol Consortium. involved in pain perception, and the catechol-O-methyl- transferase (COMT) gene is associated with multiple sorts Electronic supplementary material The online version of this article (doi:10.1007/s11136-014-0656-1) contains supplementary of pain. The neurotransmitter and neuroplasticity theories material, which is available to authorized users. -

Upregulation of the Transcription Factor TFAP2D Is Associated With
Fraune et al. Molecular Medicine (2020) 26:24 Molecular Medicine https://doi.org/10.1186/s10020-020-00148-4 RESEARCH ARTICLE Open Access Upregulation of the transcription factor TFAP2D is associated with aggressive tumor phenotype in prostate cancer lacking the TMPRSS2:ERG fusion Christoph Fraune1†, Luisa Harms1†, Franziska Büscheck1, Doris Höflmayer1, Maria Christina Tsourlakis1, Till S. Clauditz1, Ronald Simon1* , Katharina Möller1, Andreas M. Luebke1, Christina Möller-Koop1, Stefan Steurer1, Claudia Hube-Magg1, Guido Sauter1, Sören Weidemann1, Patrick Lebok1, David Dum1, Simon Kind1, Sarah Minner1, Jakob R. Izbicki2, Thorsten Schlomm3, Hartwig Huland4, Hans Heinzer4, Eike Burandt1, Alexander Haese4, Markus Graefen4 and Cornelia Schroeder2 Abstract Background: TFAP2D is a transcription factor important for modulating gene expression in embryogenesis. Its expression and prognostic role in prostate cancer has not been evaluated. Methods: Therefore, a tissue microarray containing 17,747 prostate cancer specimens with associated pathological, clinical, and molecular data was analyzed by immunohistochemistry to assess the role of TFAP2D. Results: TFAP2D expression was typically increased in prostate cancer as compared to adjacent non-neoplastic glands. TFAP2D staining was considered negative in 24.3% and positive in 75.7% of 13,545 interpretable cancers. TFAP2D staining was significantly linked to advanced tumor stage, high classical and quantitative Gleason grade, lymph node metastasis, and a positive surgical margin (p ≤ 0.0045). TFAP2D positivity was more common in ERG fusion positive (88.7%) than in ERG negative cancers (66.8%; p < 0.0001). Subset analyses in 3776 cancers with and 4722 cancers without TMPRSS2:ERG fusion revealed that associations with tumor phenotype and patient outcome were largely driven by the subset of ERG negative tumors. -

Prioritization of Metabolic Genes As Novel Therapeutic Targets
bioRxiv preprint doi: https://doi.org/10.1101/515403; this version posted January 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 Prioritization of metabolic genes as novel therapeutic targets 2 in estrogen-receptor negative breast tumors using multi-omics data and text mining 3 4 Dinesh Kumar Barupal#,1, Bei Gao#, 1, Jan Budczies2, Brett S. Phinney4, Bertrand Perroud4, 5 Carsten Denkert2,3 and Oliver Fiehn*,1 6 Affiliations 7 1West Coast Metabolomics Center, University of California, Davis, CA, 95616 8 2Institute of Pathology, Charité University Hospital, Berlin, 9 3German Institute of Pathology, Philipps-University Marburg, Germany 10 4UC Davis Genome Center, University of California, Davis, CA, 95616 11 #Co-first authors and contributed equally. 12 Email address for all authors 13 Dinesh Kumar Barupal: [email protected] 14 Bei Gao: [email protected] 15 Carsten Denkert: [email protected] 16 Brett S. Phinney: [email protected] 17 Bertrand Perroud: [email protected] 18 Jan Budczies: [email protected] 19 Oliver Fiehn: [email protected] 20 21 * Corresponding Author: 22 Oliver Fiehn 23 West Coast Metabolomics Center, 24 University of California, Davis, CA, 95616 25 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/515403; this version posted January 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Co-Regulation of Hormone Receptors, Neuropeptides, and Steroidogenic Enzymes 2 Across the Vertebrate Social Behavior Network 3 4 Brent M
bioRxiv preprint doi: https://doi.org/10.1101/435024; this version posted October 4, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Co-regulation of hormone receptors, neuropeptides, and steroidogenic enzymes 2 across the vertebrate social behavior network 3 4 Brent M. Horton1, T. Brandt Ryder2, Ignacio T. Moore3, Christopher N. 5 Balakrishnan4,* 6 1Millersville University, Department of Biology 7 2Smithsonian Conservation Biology Institute, Migratory Bird Center 8 3Virginia Tech, Department of Biological Sciences 9 4East Carolina University, Department of Biology 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 1 bioRxiv preprint doi: https://doi.org/10.1101/435024; this version posted October 4, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Running Title: Gene expression in the social behavior network 2 Keywords: dominance, systems biology, songbird, territoriality, genome 3 Corresponding Author: 4 Christopher Balakrishnan 5 East Carolina University 6 Department of Biology 7 Howell Science Complex 8 Greenville, NC, USA 27858 9 [email protected] 10 2 bioRxiv preprint doi: https://doi.org/10.1101/435024; this version posted October 4, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Supplementary Tables S1-S3
Supplementary Table S1: Real time RT-PCR primers COX-2 Forward 5’- CCACTTCAAGGGAGTCTGGA -3’ Reverse 5’- AAGGGCCCTGGTGTAGTAGG -3’ Wnt5a Forward 5’- TGAATAACCCTGTTCAGATGTCA -3’ Reverse 5’- TGTACTGCATGTGGTCCTGA -3’ Spp1 Forward 5'- GACCCATCTCAGAAGCAGAA -3' Reverse 5'- TTCGTCAGATTCATCCGAGT -3' CUGBP2 Forward 5’- ATGCAACAGCTCAACACTGC -3’ Reverse 5’- CAGCGTTGCCAGATTCTGTA -3’ Supplementary Table S2: Genes synergistically regulated by oncogenic Ras and TGF-β AU-rich probe_id Gene Name Gene Symbol element Fold change RasV12 + TGF-β RasV12 TGF-β 1368519_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 42.22 5.53 75.28 1373000_at sushi-repeat-containing protein, X-linked 2 (predicted) Srpx2 19.24 25.59 73.63 1383486_at Transcribed locus --- ARE 5.93 27.94 52.85 1367581_a_at secreted phosphoprotein 1 Spp1 2.46 19.28 49.76 1368359_a_at VGF nerve growth factor inducible Vgf 3.11 4.61 48.10 1392618_at Transcribed locus --- ARE 3.48 24.30 45.76 1398302_at prolactin-like protein F Prlpf ARE 1.39 3.29 45.23 1392264_s_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 24.92 3.67 40.09 1391022_at laminin, beta 3 Lamb3 2.13 3.31 38.15 1384605_at Transcribed locus --- 2.94 14.57 37.91 1367973_at chemokine (C-C motif) ligand 2 Ccl2 ARE 5.47 17.28 37.90 1369249_at progressive ankylosis homolog (mouse) Ank ARE 3.12 8.33 33.58 1398479_at ryanodine receptor 3 Ryr3 ARE 1.42 9.28 29.65 1371194_at tumor necrosis factor alpha induced protein 6 Tnfaip6 ARE 2.95 7.90 29.24 1386344_at Progressive ankylosis homolog (mouse)