A SILAC-Based DNA Protein Interaction Screen That Identifies Candidate Binding Proteins to Functional DNA Elements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstruction of the Global Neural Crest Gene Regulatory Network in Vivo
Reconstruction of the global neural crest gene regulatory network in vivo Ruth M Williams1, Ivan Candido-Ferreira1, Emmanouela Repapi2, Daria Gavriouchkina1,4, Upeka Senanayake1, Jelena Telenius2,3, Stephen Taylor2, Jim Hughes2,3, and Tatjana Sauka-Spengler1,∗ Supplemental Material ∗Lead and corresponding author: Tatjana Sauka-Spengler ([email protected]) 1University of Oxford, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, Oxford, OX3 9DS, UK 2University of Oxford, MRC Centre for Computational Biology, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 3University of Oxford, MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 4Present Address: Okinawa Institute of Science and Technology, Molecular Genetics Unit, Onna, 904-0495, Japan A 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R1_5-6ss) log2(R1_8-10ss) log2(R1_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.92 0 r=0.99 0 r=0.96 0 r=0.99 0 r=0.96 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R2_non-NC) log2(R2_5-6ss) log2(R3_5-6ss) log2(R2_8-10ss) log2(R3_8-10ss) 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R2_5-6ss) log2(R1_8-10ss) log2(R2_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.94 0 r=0.96 0 r=0.95 0 r=0.96 0 r=0.95 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R3_non-NC) log2(R4_5-6ss) log2(R3_5-6ss) log2(R4_8-10ss) log2(R3_8-10ss) -

Alpha Actinin 4: an Intergral Component of Transcriptional
ALPHA ACTININ 4: AN INTERGRAL COMPONENT OF TRANSCRIPTIONAL PROGRAM REGULATED BY NUCLEAR HORMONE RECEPTORS By SIMRAN KHURANA Submitted in partial fulfillment of the requirements for the degree of doctor of philosophy Thesis Advisor: Dr. Hung-Ying Kao Department of Biochemistry CASE WESTERN RESERVE UNIVERSITY August, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of SIMRAN KHURANA ______________________________________________________ PhD candidate for the ________________________________degree *. Dr. David Samols (signed)_______________________________________________ (chair of the committee) Dr. Hung-Ying Kao ________________________________________________ Dr. Edward Stavnezer ________________________________________________ Dr. Leslie Bruggeman ________________________________________________ Dr. Colleen Croniger ________________________________________________ ________________________________________________ May 2011 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS LIST OF TABLES vii LIST OF FIGURES viii ACKNOWLEDEMENTS xii LIST OF ABBREVIATIONS xiii ABSTRACT 1 CHAPTER 1: INTRODUCTION Family of Nuclear Receptors 3 Mechanism of transcriptional regulation by co-repressors and co-activators 8 Importance of LXXLL motif of co-activators in NR mediated transcription 12 Cyclic recruitment of co-regulators on the target promoters 15 Actin and actin related proteins (ABPs) in transcription -

Gene PMID WBS Locus ABR 26603386 AASDH 26603386
Supplementary material J Med Genet Gene PMID WBS Locus ABR 26603386 AASDH 26603386 ABCA1 21304579 ABCA13 26603386 ABCA3 25501393 ABCA7 25501393 ABCC1 25501393 ABCC3 25501393 ABCG1 25501393 ABHD10 21304579 ABHD11 25501393 yes ABHD2 25501393 ABHD5 21304579 ABLIM1 21304579;26603386 ACOT12 25501393 ACSF2,CHAD 26603386 ACSL4 21304579 ACSM3 26603386 ACTA2 25501393 ACTN1 26603386 ACTN3 26603386;25501393;25501393 ACTN4 21304579 ACTR1B 21304579 ACVR2A 21304579 ACY3 19897463 ACYP1 21304579 ADA 25501393 ADAM12 21304579 ADAM19 25501393 ADAM32 26603386 ADAMTS1 25501393 ADAMTS10 25501393 ADAMTS12 26603386 ADAMTS17 26603386 ADAMTS6 21304579 ADAMTS7 25501393 ADAMTSL1 21304579 ADAMTSL4 25501393 ADAMTSL5 25501393 ADCY3 25501393 ADK 21304579 ADRBK2 25501393 AEBP1 25501393 AES 25501393 AFAP1,LOC84740 26603386 AFAP1L2 26603386 AFG3L1 21304579 AGAP1 26603386 AGAP9 21304579 Codina-Sola M, et al. J Med Genet 2019; 56:801–808. doi: 10.1136/jmedgenet-2019-106080 Supplementary material J Med Genet AGBL5 21304579 AGPAT3 19897463;25501393 AGRN 25501393 AGXT2L2 25501393 AHCY 25501393 AHDC1 26603386 AHNAK 26603386 AHRR 26603386 AIDA 25501393 AIFM2 21304579 AIG1 21304579 AIP 21304579 AK5 21304579 AKAP1 25501393 AKAP6 21304579 AKNA 21304579 AKR1E2 26603386 AKR7A2 21304579 AKR7A3 26603386 AKR7L 26603386 AKT3 21304579 ALDH18A1 25501393;25501393 ALDH1A3 21304579 ALDH1B1 21304579 ALDH6A1 21304579 ALDOC 21304579 ALG10B 26603386 ALG13 21304579 ALKBH7 25501393 ALPK2 21304579 AMPH 21304579 ANG 21304579 ANGPTL2,RALGPS1 26603386 ANGPTL6 26603386 ANK2 21304579 ANKMY1 26603386 ANKMY2 -

Genes of the Escherichia Coli Pur Regulon Are Negatively Controlled
JOURNAL OF BACTERIOLOGY, Aug. 1990, p. 4555-4562 Vol. 172, No. 8 0021-9193/90/084555-08$02.00/0 Copyright © 1990, American Society for Microbiology Genes of the Escherichia coli pur Regulon Are Negatively Controlled by a Repressor-Operator Interactiont BIN HE,' ALLAN SHIAU,' KANG YELL CHOI,' HOWARD ZALKIN,1* AND JOHN M. SMITH2 Department ofBiochemistry, Purdue University, West Lafayette, Indiana 47907,' and Seattle Biomedical Research Institute, Seattle, Washington 981092 Received 1 March 1990/Accepted 22 May 1990 Fusions of lacZ were constructed to genes in each of the loci involved in de novo synthesis of IMP. The expression of each pur-lacZ fusion was determined in isogenic purR and purR+ strains. These measurements indicated 5- to 17-fold coregulation of genes purF, purHD, purC, purMN, purL, and purEK and thus confirm the existence of a pur regulon. Gene purB, which encodes an enzyme involved in synthesis of IMP and in the AMP branch of the pathway, was not regulated by purR. Each locus of the pur regulon contains a 16-base-pair conserved operator sequence that overlaps with the promoter. The purR product, purine repressor, was shown to bind specifically to each operator. Thus, binding of repressor to each operator of pur regulon genes negatively coregulates expression. In all organisms there are 10 steps for de novo synthesis of more, the effector molecules that act as coregulators have IMP, the first purine nucleotide intermediate in the pathway. not been identified. Nucleotides have been assumed to be IMP is a branch point metabolite which is converted to the effector molecules, but the purine bases hypoxanthine adenine and quanine nucleotides (Fig. -

Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases
Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Olivares, Ana Maria, Oscar Andrés Moreno-Ramos, and Neena B. Haider. 2015. “Role of Nuclear Receptors in Central Nervous System Development and Associated Diseases.” Journal of Experimental Neuroscience 9 (Suppl 2): 93-121. doi:10.4137/JEN.S25480. http:// dx.doi.org/10.4137/JEN.S25480. Published Version doi:10.4137/JEN.S25480 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:27320246 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Journal name: Journal of Experimental Neuroscience Journal type: Review Year: 2015 Volume: 9(S2) Role of Nuclear Receptors in Central Nervous System Running head verso: Olivares et al Development and Associated Diseases Running head recto: Nuclear receptors development and associated diseases Supplementary Issue: Molecular and Cellular Mechanisms of Neurodegeneration Ana Maria Olivares1, Oscar Andrés Moreno-Ramos2 and Neena B. Haider1 1Department of Ophthalmology, Schepens Eye Research Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA. 2Departamento de Ciencias Biológicas, Facultad de Ciencias, Universidad de los Andes, Bogotá, Colombia. ABSTRACT: The nuclear hormone receptor (NHR) superfamily is composed of a wide range of receptors involved in a myriad of important biological processes, including development, growth, metabolism, and maintenance. -
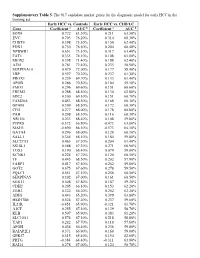
Supplementary Table 5. the 917 Candidate Marker Genes for the Diagnostic Model for Early HCC in the Training Set
Supplementary Table 5. The 917 candidate marker genes for the diagnostic model for early HCC in the training set. Early HCC vs. Controls Early HCC vs. CHB/LC Gene Coefficient a AUC b Coefficient a AUC b SOX9 0.722 81.30% 0.211 63.50% EVC 0.703 76.20% 0.314 68.30% CHST9 0.398 75.50% 0.150 62.40% PDX1 0.730 76.50% 0.204 60.40% NPBWR1 0.651 73.10% 0.317 63.40% FAT1 0.335 74.10% 0.108 61.00% MEIS2 0.398 71.40% 0.188 62.40% A2M 0.761 72.40% 0.235 58.90% SERPINA10 0.479 72.00% 0.177 58.40% LBP 0.597 70.20% 0.237 61.30% PROX1 0.239 69.70% 0.133 61.40% APOB 0.286 70.50% 0.104 59.10% FMO3 0.296 69.60% 0.151 60.60% FREM2 0.288 68.50% 0.130 62.80% SDC2 0.300 69.10% 0.151 60.70% FAM20A 0.453 68.50% 0.168 60.30% GPAM 0.309 68.50% 0.172 60.30% CFH 0.277 68.00% 0.178 60.80% PAH 0.208 68.30% 0.116 60.30% NR1H4 0.233 68.40% 0.108 59.80% PTPRS -0.572 66.80% -0.473 63.00% SIAH3 -0.690 66.10% -0.573 64.10% GATA4 0.296 68.00% 0.128 60.10% SALL1 0.344 68.10% 0.184 59.80% SLC27A5 0.463 67.30% 0.204 61.40% SS18L1 0.588 67.30% 0.271 60.90% TOX3 0.190 68.40% 0.079 59.00% KCNK1 0.224 67.70% 0.120 60.10% TF 0.445 68.50% 0.202 57.90% FARP1 0.417 67.50% 0.252 59.80% GOT2 0.675 67.60% 0.278 59.50% PQLC1 0.651 67.10% 0.258 60.50% SERPINA5 0.302 67.00% 0.161 60.50% SOX13 0.508 67.80% 0.187 59.30% CDH2 0.205 66.10% 0.153 62.20% ITIH2 0.322 66.20% 0.252 62.20% ADIG 0.443 65.20% 0.399 63.80% HSD17B6 0.524 67.20% 0.237 59.60% IL21R -0.451 65.90% -0.321 61.70% A1CF 0.255 67.10% 0.139 58.70% KLB 0.507 65.90% 0.383 61.20% SLC10A1 0.574 67.10% 0.218 58.80% YAP1 0.282 67.70% 0.118 -

Adenylosuccinate Lyase of Bacillus Subtilis Regulates the Activity of The
Proc. Nati. Acad. Sci. USA Vol. 89, pp. 5389-5392, June 1992 Biochemistry Adenylosuccinate lyase of Bacillus subtilis regulates the activity of the glutamyl-tRNA synthetase (purB/adenylosuccinate AMP-lyase/glutamate-tRNA ligase) NATHALIE GENDRON, ROCK BRETON, NATHALIE CHAMPAGNE, AND JACQUES LAPOINTE* DUpartement de Biochimie, Facultd des Sciences et de Gdnie, Universitt Laval, Qudbec, Canada, GlK 7P4 Communicated by H. E. Umbarger, February 21, 1992 (received for review November 27, 1991) ABSTRACT In Bacillus subtilis, the glutamyl-tRNA syn- Table 1. Bacterial strains used thetase [L-glutamate:tRNAGlU ligase (AMP-forming), EC Strain Genotype Ref. 6.1.1.17] is copurified with a polypeptide of Mr 46,000 that influences its affinity for its substrates and increases its ther- E. coli DH5a 080dlacZAM15, endAI, recAl, 11 mostability. The gene encoding this regulatory factor was hsdR17 (RK-, MK+), supE44, cloned with the aid of a 41-mer oligonucleotide probe corre- thi-l, A-, gyrA, relAI, F-, sponding to the amino acid sequence of an NH2-terminal A&(IacZYA-argF) U169 ofthis factor. The nucleotide sequence ofthis gene and E. coli JK268 F-, trpE61, trpA62, tna-5, 12 segment purB58, A- the physical map of the 1475-base-pair fragment on which it E. coli JM101 supE thi-1 A(lac-proAB) 13 was cloned are identical to those of purB, which encodes the F'[traD36 proAB+ Iaclq adenylosuccinate lyase (adenylosuccinate AMP-lyase, EC lacZAM15] 4.3.2.2), an enzyme involved in the de novo synthesis of B. subtilis 1A1* trpC2 14 purines. This gene complements the purB mutation of Esche- richia coli JK268, and its presence on a multicopy plasmid behind the trc promoter in the purB- strain gives an adenylo- extract (10). -

Predicting Transcription Factor Specificity with All-Atom Models Sahand Jamal Rahi1, Peter Virnau2,*, Leonid A
Published online 1 October 2008 Nucleic Acids Research, 2008, Vol. 36, No. 19 6209–6217 doi:10.1093/nar/gkn589 Predicting transcription factor specificity with all-atom models Sahand Jamal Rahi1, Peter Virnau2,*, Leonid A. Mirny1,3 and Mehran Kardar1 1Department of Physics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA, 2Staudinger Weg 7, Institut fu¨ r Physik, 55099 Mainz, Germany and 3Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA Downloaded from https://academic.oup.com/nar/article/36/19/6209/2410008 by guest on 02 October 2021 Received May 4, 2008; Revised August 29, 2008; Accepted September 2, 2008 ABSTRACT needs to have a set of known sites bound by the protein. Given these known sites, the parameters are inferred using The binding of a transcription factor (TF) to a DNA either a widely used Berg–von Hippel approximation (1), operator site can initiate or repress the expression or by other recently proposed methods (2–4). This consti- of a gene. Computational prediction of sites recog- tutes a physical basis for many widely used bioinformatics nized by a TF has traditionally relied upon knowl- techniques that rely on a particular form of the energy edge of several cognate sites, rather than an ab function known as a position-specific weight matrix initio approach. Here, we examine the possibility (PWM). of using structure-based energy calculations that All these methods require a priori knowledge of the require no knowledge of bound sites but rather sites (or at least longer sequences containing these sites) start with the structure of a protein–DNA complex. -

Anti-Testicular Receptor 2 (TR2) (Rabbit) Antibody - 100-401-E45
Anti-Testicular Receptor 2 (TR2) (Rabbit) Antibody - 100-401-E45 Code: 100-401-E45 Size: 100 µL Product Description: Anti-Testicular Receptor 2 (TR2) (Rabbit) Antibody - 100-401-E45 Concentration: Titrated value sufficient to run approximately 10 mini blots. PhysicalState: Liquid Label Unconjugated Host Rabbit Gene Name NR2C1 Species Reactivity mouse, human, rat Storage Condition Store vial at -20° C prior to opening. This product is stable at 4° C as an undiluted liquid. For extended storage, aliquot contents and freeze at -20° C or below. Avoid cycles of freezing and thawing. Dilute only prior to immediate use. Synonyms Nuclear receptor subfamily 2 group C member 1, Orphan nuclear receptor TR2, mTR2 Application Note Anti-Testicular Receptor 2 (Rabbit) antibody is suitable for use in Western Blots. Anti-Testicular Receptor 2 antibodies are specific for the ~64 kDa TR2 protein in Western blots of testes and nuclear extracts from MEL cell lines. Researchers should determine optimal titers for applications that are not stated below. Background TR2 antibody detects Testicular receptor 2 (TR2), which is a member of the orphan nuclear receptor family. It is widely expressed at a low level throughout the adult testis. TR2 represses transcription and binds DNA directly interacting with HDAC3 and HDAC4 via DNA-binding domains. TR2 has also been implicated in regulation of estrogen receptor activity in mammary glands. In addition, TR2 has recently been shown to form a heterodimer with TR4 that can bind to the direct repeat 6 element of the hepatitis B virus (HBV) enhancer II region thus suppressing HBV gene expression. -

PURB Antibody (C-Term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP12051B
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 PURB Antibody (C-term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP12051B Specification PURB Antibody (C-term) - Product Information Application IF, WB, FC,E Primary Accession Q96QR8 Other Accession Q68A21, O35295, NP_150093.1 Reactivity Human Predicted Mouse, Rat Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Antigen Region 260-289 PURB Antibody (C-term) - Additional Information Gene ID 5814 Other Names Fluorescent image of A549 cell stained with Transcriptional activator protein Pur-beta, PURB Antibody Purine-rich element-binding protein B, PURB (C-term)(Cat#AP12051b).A549 cells were fixed with 4% PFA (20 min), permeabilized Target/Specificity with Triton X-100 (0.1%, 10 min), then This PURB antibody is generated from incubated with PURB primary antibody (1:25, rabbits immunized with a KLH conjugated 1 h at 37℃). For secondary antibody, Alexa synthetic peptide between 260-289 amino Fluor® 488 conjugated donkey anti-rabbit acids from the C-terminal region of human antibody (green) was used (1:400, 50 min at PURB. 37℃).Cytoplasmic actin was counterstained with Alexa Fluor® 555 (red) conjugated Dilution IF~~1:10~50 Phalloidin (7units/ml, 1 h at 37℃).PURB WB~~1:1000 immunoreactivity is localized to Nucleus FC~~1:10~50 significantly. Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. Storage Maintain refrigerated at 2-8°C for up to 2 weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw cycles. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice.