Downloaded a Meta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
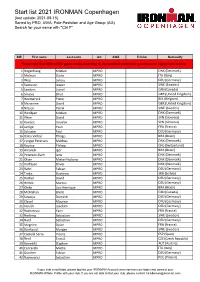
Start List 2021 IRONMAN Copenhagen (Last Update: 2021-08-15) Sorted by PRO, AWA, Pole Posistion and Age Group (AG) Search for Your Name with "Ctrl F"
Start list 2021 IRONMAN Copenhagen (last update: 2021-08-15) Sorted by PRO, AWA, Pole Posistion and Age Group (AG) Search for your name with "Ctrl F" BIB First name Last name AG AWA TriClub Nationallty Please note that BIBs will be given onsite according to the selected swim time you choose in registration oniste. 1 Hogenhaug Kristian MPRO DNK (Denmark) 2 Molinari Giulio MPRO ITA (Italy) 3 Wojt Lukasz MPRO DEU (Germany) 4 Svensson Jesper MPRO SWE (Sweden) 5 Sanders Lionel MPRO CAN (Canada) 6 Smales Elliot MPRO GBR (United Kingdom) 7 Heemeryck Pieter MPRO BEL (Belgium) 8 Mcnamee David MPRO GBR (United Kingdom) 9 Nilsson Patrik MPRO SWE (Sweden) 10 Hindkjær Kristian MPRO DNK (Denmark) 11 Plese David MPRO SVN (Slovenia) 12 Kovacic Jaroslav MPRO SVN (Slovenia) 14 Jarrige Yvan MPRO FRA (France) 15 Schuster Paul MPRO DEU (Germany) 16 Dário Vinhal Thiago MPRO BRA (Brazil) 17 Lyngsø Petersen Mathias MPRO DNK (Denmark) 18 Koutny Philipp MPRO CHE (Switzerland) 19 Amorelli Igor MPRO BRA (Brazil) 20 Petersen-Bach Jens MPRO DNK (Denmark) 21 Olsen Mikkel Hojborg MPRO DNK (Denmark) 22 Korfitsen Oliver MPRO DNK (Denmark) 23 Rahn Fabian MPRO DEU (Germany) 24 Trakic Strahinja MPRO SRB (Serbia) 25 Rother David MPRO DEU (Germany) 26 Herbst Marcus MPRO DEU (Germany) 27 Ohde Luis Henrique MPRO BRA (Brazil) 28 McMahon Brent MPRO CAN (Canada) 29 Sowieja Dominik MPRO DEU (Germany) 30 Clavel Maurice MPRO DEU (Germany) 31 Krauth Joachim MPRO DEU (Germany) 32 Rocheteau Yann MPRO FRA (France) 33 Norberg Sebastian MPRO SWE (Sweden) 34 Neef Sebastian MPRO DEU (Germany) 35 Magnien Dylan MPRO FRA (France) 36 Björkqvist Morgan MPRO SWE (Sweden) 37 Castellà Serra Vicenç MPRO ESP (Spain) 38 Řenč Tomáš MPRO CZE (Czech Republic) 39 Benedikt Stephen MPRO AUT (Austria) 40 Ceccarelli Mattia MPRO ITA (Italy) 41 Günther Fabian MPRO DEU (Germany) 42 Najmowicz Sebastian MPRO POL (Poland) If your club is not listed, please log into your IRONMAN Account (www.ironman.com/login) and connect your IRONMAN Athlete Profile with your club. -

Legacy Image
5-(948-~gblb SOWARE ENGINEERING LABORATORY SERIES SEL-95-004 PROCEEDINGS OF THE TWENTIETH ANNUAL SOFTWARE ENGINEERING WORKSHOP DECEMBER 1995 National Aeronautics and Space Administration Goddard Space Flight Center Greenbelt, Maryland 20771 Proceedings of the Twentieth Annual Software Engineering Workshop November 29-30,1995 GODDARD SPACE FLIGHT CENTER Greenbelt, Maryland The Software Engineering Laboratory (SEL) is an organization sponsored by the National Aeronautics and Space AdministratiodGoddard Space Flight Center (NASAIGSFC) and created to investigate the effectiveness of software engineering technologies when applied to the development of applications software. The SEL was created in 1976 and has three primary organizational members: NASAIGSFC, Software Engineering Branch The University of Maryland, Department of Computer Science .I Computer Sciences Corporation, Software Engineering Operation The goals of the SEL are (1) to understand the software development process in the GSFC environment; (2) to measure the effects of various methodologies, tools, and models on this process; and (3) to identifjr and then to apply successful development practices. The activities, findings, and recommendations of the SEL are recorded in the Software Engineering Laboratory Series, a continuing series of reports that includes this document. Documents from the Software Engineering Laboratory Series can be obtained via the SEL homepage at: or by writing to: Software Engineering Branch Code 552 Goddard Space Flight Center Greenbelt, Maryland 2077 1 SEW Proceedings iii The views and findings expressed herein are those of, the authors and presenters and do not necessarily represent the views, estimates, or policies of the SEL. All material herein is reprinted as submitted by authors and presenters, who are solely responsible for compliance with any relevant copyright, patent, or other proprietary restrictions. -

Reconstruction of the Global Neural Crest Gene Regulatory Network in Vivo
Reconstruction of the global neural crest gene regulatory network in vivo Ruth M Williams1, Ivan Candido-Ferreira1, Emmanouela Repapi2, Daria Gavriouchkina1,4, Upeka Senanayake1, Jelena Telenius2,3, Stephen Taylor2, Jim Hughes2,3, and Tatjana Sauka-Spengler1,∗ Supplemental Material ∗Lead and corresponding author: Tatjana Sauka-Spengler ([email protected]) 1University of Oxford, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, Oxford, OX3 9DS, UK 2University of Oxford, MRC Centre for Computational Biology, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 3University of Oxford, MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 4Present Address: Okinawa Institute of Science and Technology, Molecular Genetics Unit, Onna, 904-0495, Japan A 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R1_5-6ss) log2(R1_8-10ss) log2(R1_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.92 0 r=0.99 0 r=0.96 0 r=0.99 0 r=0.96 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R2_non-NC) log2(R2_5-6ss) log2(R3_5-6ss) log2(R2_8-10ss) log2(R3_8-10ss) 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R2_5-6ss) log2(R1_8-10ss) log2(R2_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.94 0 r=0.96 0 r=0.95 0 r=0.96 0 r=0.95 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R3_non-NC) log2(R4_5-6ss) log2(R3_5-6ss) log2(R4_8-10ss) log2(R3_8-10ss) -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Gene PMID WBS Locus ABR 26603386 AASDH 26603386
Supplementary material J Med Genet Gene PMID WBS Locus ABR 26603386 AASDH 26603386 ABCA1 21304579 ABCA13 26603386 ABCA3 25501393 ABCA7 25501393 ABCC1 25501393 ABCC3 25501393 ABCG1 25501393 ABHD10 21304579 ABHD11 25501393 yes ABHD2 25501393 ABHD5 21304579 ABLIM1 21304579;26603386 ACOT12 25501393 ACSF2,CHAD 26603386 ACSL4 21304579 ACSM3 26603386 ACTA2 25501393 ACTN1 26603386 ACTN3 26603386;25501393;25501393 ACTN4 21304579 ACTR1B 21304579 ACVR2A 21304579 ACY3 19897463 ACYP1 21304579 ADA 25501393 ADAM12 21304579 ADAM19 25501393 ADAM32 26603386 ADAMTS1 25501393 ADAMTS10 25501393 ADAMTS12 26603386 ADAMTS17 26603386 ADAMTS6 21304579 ADAMTS7 25501393 ADAMTSL1 21304579 ADAMTSL4 25501393 ADAMTSL5 25501393 ADCY3 25501393 ADK 21304579 ADRBK2 25501393 AEBP1 25501393 AES 25501393 AFAP1,LOC84740 26603386 AFAP1L2 26603386 AFG3L1 21304579 AGAP1 26603386 AGAP9 21304579 Codina-Sola M, et al. J Med Genet 2019; 56:801–808. doi: 10.1136/jmedgenet-2019-106080 Supplementary material J Med Genet AGBL5 21304579 AGPAT3 19897463;25501393 AGRN 25501393 AGXT2L2 25501393 AHCY 25501393 AHDC1 26603386 AHNAK 26603386 AHRR 26603386 AIDA 25501393 AIFM2 21304579 AIG1 21304579 AIP 21304579 AK5 21304579 AKAP1 25501393 AKAP6 21304579 AKNA 21304579 AKR1E2 26603386 AKR7A2 21304579 AKR7A3 26603386 AKR7L 26603386 AKT3 21304579 ALDH18A1 25501393;25501393 ALDH1A3 21304579 ALDH1B1 21304579 ALDH6A1 21304579 ALDOC 21304579 ALG10B 26603386 ALG13 21304579 ALKBH7 25501393 ALPK2 21304579 AMPH 21304579 ANG 21304579 ANGPTL2,RALGPS1 26603386 ANGPTL6 26603386 ANK2 21304579 ANKMY1 26603386 ANKMY2 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

TMEM106B in Humans and Vac7 and Tag1 in Yeast Are Predicted to Be Lipid Transfer Proteins
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.12.435176; this version posted March 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. TMEM106B in humans and Vac7 and Tag1 in yeast are predicted to be lipid transfer proteins Tim P. Levine* UCL Institute of Ophthalmology, 11-43 Bath Street, London EC1V 9EL, United Kingdom. ORCID 0000-0002-7231-0775 *Corresponding author and lead contact: [email protected] Data availability statement: The data that support this study are freely available in Harvard Dataverse at https://dataverse.harvard.edu/dataverse/LEA_2. Acknowledgements: work was funded by the Higher Education Funding Council for England and the NIHR Moorfields Biomedical Research Centre Conflict of interest disclosure: the author declares that there is no conflict of interest Keywords: Structural bioinformatics, Lipid transfer protein, LEA_2, TMEM106B, Vac7, YLR173W, Endosome, Lysosome Running Title: TMEM106B & Vac7: lipid transfer proteins 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.12.435176; this version posted March 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract TMEM106B is an integral membrane protein of late endosomes and lysosomes involved in neuronal function, its over-expression being associated with familial frontotemporal lobar degeneration, and under-expression linked to hypomyelination. It has also been identified in multiple screens for host proteins required for productive SARS-CoV2 infection. Because standard approaches to understand TMEM106B at the sequence level find no homology to other proteins, it has remained a protein of unknown function. -

Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement
School of Natural Sciences and Mathematics Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement UT Dallas Author(s): Michael Q. Zhang Rights: CC BY 4.0 (Attribution) ©2017 The Authors Citation: Liu, Zehua, Huazhe Lou, Kaikun Xie, Hao Wang, et al. 2017. "Reconstructing cell cycle pseudo time-series via single-cell transcriptome data." Nature Communications 8, doi:10.1038/s41467-017-00039-z This document is being made freely available by the Eugene McDermott Library of the University of Texas at Dallas with permission of the copyright owner. All rights are reserved under United States copyright law unless specified otherwise. File name: Supplementary Information Description: Supplementary figures, supplementary tables, supplementary notes, supplementary methods and supplementary references. CCNE1 CCNE1 CCNE1 CCNE1 36 40 32 34 32 35 30 32 28 30 30 28 28 26 24 25 Normalized Expression Normalized Expression Normalized Expression Normalized Expression 26 G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage CCNE1 CCNE1 CCNE1 CCNE1 40 32 40 40 35 30 38 30 30 28 36 25 26 20 20 34 Normalized Expression Normalized Expression Normalized Expression 24 Normalized Expression G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Supplementary Figure 1 | High stochasticity of single-cell gene expression means, as demonstrated by relative expression levels of gene Ccne1 using the mESC-SMARTer data. For every panel, 20 sample cells were randomly selected for each of the three stages, followed by plotting the mean expression levels at each stage. -

A Bootstrap-Based Regression Method for Comprehensive Discovery of Differential Gene Expressions: an Application to the Osteoporosis Study
A bootstrap-based regression method for comprehensive discovery of differential gene expressions: an application to the osteoporosis study Yan Lu1,2, Yao-Zhong Liu3, Peng–Yuan Liu2, Volodymyr Dvornyk4, and Hong–Wen Deng1,3 1. College of Life Sciences and Bioengineering, Beijing Jiaotong University, Beijing 100044, P. R. China 2. Department of Physiology and the Cancer Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA 3. Department of Biostatistics and Bioinformatics & Center for Bioinformatics and Genomics, Tulane University School of Public Health, New Orleans, LA 70112, USA 4. School of Biological Sciences, University of Hong Kong, Pokfulam Rd., Pokfulam, Hong Kong, P.R. China Running title: Bootstrap-based regression method for microarray analysis Key words: microarray, bootstrap, regression, osteoporosis Corresponding author: Hong–Wen Deng, Ph. D. Department of Biostatistics and Bioinformatics & Center for Bioinformatics and Genomics, Tulane University School of Public Health, 1440 Canal Street, Suite 2001, New Orleans, LA 70112 Tel: 504-988-1310 Email: [email protected] 1 Abstract A common purpose of microarray experiments is to study the variation in gene expression across the categories of an experimental factor such as tissue types and drug treatments. However, it is not uncommon that the studied experimental factor is a quantitative variable rather than categorical variable. Loss of information would occur by comparing gene-expression levels between groups that are factitiously defined according to the quantitative threshold values of an experimental factor. Additionally, lack of control for some sensitive clinical factors may bring serious false positive or negative findings. In the present study, we described a bootstrap-based regression method for analyzing gene expression data from the non-categorical microarray experiments. -

Thèse De Doctorat De L'université Paris-Saclay Préparée À L'ecole
1 NNT : 2017SACLX031 Thèse de doctorat de l’Université Paris-Saclay préparée à l’Ecole Polytechnique Ecole doctorale n◦573 Interfaces : approches interdisciplinaires / fondements, applications et innovation Spécialité de doctorat : Informatique par Mme. Alice Héliou Analyse des séquences génomiques : Identification des ARNs circulaires et calcul de l’information négative Thèse présentée et soutenue à l’Ecole Polytechnique, le 10 juillet 2017. Composition du Jury : M. Alain Denise Professeur (Président du jury) Université Paris-Sud Mme. Christine Gaspin Professeur (Rapporteur) INRA Toulouse M. Gabriele Fici Professeur associé (Rapporteur) Université de Palerme (Italie) M Thierry Lecroq Professeur (Examinateur) Université de Rouen Mme. Claire Toffano-Nioche Chargé de recherche (Examinatrice) Université Paris Sud M. Mikael Salson Maitre de conférences (Examinateur) Université Lille 1 Mme. Mireille Régnier Professeur (Directrice de thèse) Ecole Polytechnique M. Hubert Becker Maitre de conférences (Co-directeur de thèse) UPMC À mes grands parents, Pour m’avoir ouvert les yeux sur la chance que sont les études. À mes parents, Pour m’avoir toujours soutenue et donné confiance en moi, Merci. Remerciements Je tiens dans un premier temps à remercier mes directeurs de thèse, Mireille Régnier et Hubert Becker sans qui rien de tout cela n’aurait été possible. Christine Gaspin et Gabriele Fici ont accepté d’être rapporteurs de cette thèse, je les remercie sincèrement d’avoir pris le temps de lire attentivement le manuscrit. Je souhaite également remercier Alain Denise, Thierry Lecroq, Claire Toffano- Nioche et Mikael Salson d’avoir accepté de faire partie de mon jury pour la soute- nance. La première personne qu’il m’est important de remercier est Laurent Mouchard. -

Statistical and Bioinformatic Analysis of Hemimethylation Patterns in Non-Small Cell Lung Cancer
Statistical and Bioinformatic Analysis of Hemimethylation Patterns in Non-Small Cell Lung Cancer Shuying Sun ( [email protected] ) Texas State University San Marcos https://orcid.org/0000-0003-3974-6996 Austin Zane Texas A&M University College Station Carolyn Fulton Schreiner University Jasmine Philipoom Case Western Reserve University Research article Keywords: Methylation, Hemimethylation, Lung Cancer, Bioinformatics, Epigenetics Posted Date: October 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-17794/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on March 12th, 2021. See the published version at https://doi.org/10.1186/s12885-021-07990-7. Page 1/29 Abstract Background: DNA methylation is an epigenetic event involving the addition of a methyl-group to a cytosine-guanine base pair (i.e., CpG site). It is associated with different cancers. Our research focuses on studying non- small cell lung cancer hemimethylation, which refers to methylation occurring on only one of the two DNA strands. Many studies often assume that methylation occurs on both DNA strands at a CpG site. However, recent publications show the existence of hemimethylation and its signicant impact. Therefore, it is important to identify cancer hemimethylation patterns. Methods: In this paper, we use the Wilcoxon signed rank test to identify hemimethylated CpG sites based on publicly available non-small cell lung cancer methylation sequencing data. We then identify two types of hemimethylated CpG clusters, regular and polarity clusters, and genes with large numbers of hemimethylated sites. -

DEAD-Box Helicase 27 Enhances Stem Cell-Like Properties with Poor Prognosis in Breast Cancer
DEAD-Box Helicase 27 Enhances Stem Cell-Like Properties With Poor Prognosis in Breast Cancer Shan Li The First Aliated Hospital of China Medical University Jinfei Ma The First Aliated Hospital of China Medical University Ang Zheng The First Aliated Hospital of China Medical University Xinyue Song China Medical University Si Chen China Medical University Feng Jin ( [email protected] ) The First Aliated Hospital of China Medical University https://orcid.org/0000-0002-0325-5362 Research Article Keywords: DEAD-box helicase 27 (DDX27), Breast cancer, Stem cell-like properties, Prognosis Posted Date: June 7th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-521379/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Journal of Translational Medicine on August 6th, 2021. See the published version at https://doi.org/10.1186/s12967-021-03011-0. Page 1/22 Abstract Background Although the rapid development of diagnosis and treatment has improved prognosis in early breast cancer, challenges from different therapy response remain due to breast cancer heterogeneity. DEAD-box helicase 27 (DDX27) had been proved to inuence ribosome biogenesis and identied as a promoter in gastric and colorectal cancer associated with stem cell-like properties, while the impact of DDX27 on breast cancer prognosis and biological functions is unclear. We aimed to explore the inuence of DDX27 on stem cell-like properties and prognosis in breast cancer. Methods The expression of DDX27 was evaluated in 24 pairs of fresh breast cancer and normal tissue by western blot.