The Use of Bacteroides Genetic Markers to Identify
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Potential Role of Microbiome in Oncogenesis, Outcome Prediction
Oral Oncology 99 (2019) 104453 Contents lists available at ScienceDirect Oral Oncology journal homepage: www.elsevier.com/locate/oraloncology Review Potential role of microbiome in oncogenesis, outcome prediction and therapeutic targeting for head and neck cancer T ⁎ Ester Orlandia,b, , Nicola Alessandro Iacovellib, Vincenzo Tombolinic, Tiziana Rancatid, Antonella Polimenie, Loris De Ceccof, Riccardo Valdagnia,d,g, Francesca De Felicec a Department of Radiotherapy 1, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy b Department of Radiotherapy 2, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy c Department of Radiotherapy, Policlinico Umberto I, “Sapienza” University of Rome, Rome, Italy d Prostate Cancer Program, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy e Department of Oral and Maxillo Facial Sciences, Policlinico Umberto I, “Sapienza” University of Rome, Italy f Integrated Biology Platform, Department of Applied Research and Technology Development, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy g Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy ARTICLE INFO ABSTRACT Keywords: In the last decade, human microbiome research is rapidly growing involving several fields of clinical medicine Head and neck cancer and population health. Although the microbiome seems to be linked to all sorts of diseases, cancer has the Biomarkers biggest potential to be investigated. Microbiome Following the publication of the National Institute of Health - Human Microbiome Project (NIH-HMP), the Microbiota link between Head and Neck Cancer (HNC) and microbiome seems to be a fast-moving field in research area. Oncogenesis However, robust evidence-based literature is still quite scarce. Nevertheless the relationship between oral mi- Radiotherapy Immunotherapy crobiome and HNC could have important consequences for prevention and early detection of this type of tumors. -

3. Jedna Z Krvných Skupín Systému AB0; 4. Skr. Bukálny. B – Symbo
B – 1. symbol pre bel; 2. chem. značka prvku →bór; 3. jedna z krvných skupín systému AB0; 4. skr. bukálny. B – symbol pre hustotu magnetického toku. B. – skr. pre →Bacillus. B-ALP – skr. angl. bone alcalic phosphatase kostná alkalická fosfatáza. B-bunky – 1. syn. -bunky Langerhansových ostrovčekov; →pankreas; 2. syn. B-lymfocyty; →lymfocyty. B-komplex – multivitamínový prípravok vitamínov B. B-lymfocyty – [B podľa Fabriciovej burzy, imunol. orgánu vtákov] B bunky, druh lymfocytov, kt. sa zúčastňuje na humorálnej imunite (tvorbe protilátok) a niekt. ďalších imunitných funkciách. Povrchové molekuly sú CD 19,20. Receptorom pre antigén je membránový imunoglobulín. B-reťazec – jeden z polypeptidových reťazcov →inzulínu. B-vírus – 1. vírus →hepatitídy B.; 2. vírus zo skupiny Cercopithecine herpes virus 1. B-vlákna →nerv. b – skr. 1. pre barn; 2. skr. angl. born narodený; 3. skr. báza (genet. označenie dĺţky sekvencie nukleotidov, napr. 50 b = sekvencia 50 nukleotidov). b-vlna – pozit. kmit s vyskou amplitúdou v →elektroretinograme nasledujúci po vlne a, prejav komplexnej aktivity vrstvy bipolárnych buniek. – - – predpona označujúca 1. staršie označenie druhého atómu uhlíka v reťazci, na kt. sa pripája hlavná funkčná skupina, napr. kys. -hydroxymaslová správ. kys. 3-hydroxymaslová; 2. špecifická rotácia opticky aktívnej látky, napr. -D-glukóza; 3. orientácia exocyklického atómu al. skupiny, napr. cholest-5-en-3--ol; 4. plazmatický proteín, kt. migruje v elektroforéze v pruhu - lipoproteín; 5. člen série príbuzných chem. látok, najmä série stereoizomérov, izomérov, polymérov al. alotroických foriem, napr. -karotén; 6. -lúč. B2 – staršie označenie pre CD21. B4 – staršie označenie pre CD 19. B19 virus – ľudský parvovírus z čeľade Parvoviridae, značne rozšírený, vyvolávajúci obvykle inaparentné infekcie. -

974-Form.Pdf
California Association for Medical Laboratory Technology Distance Learning Program ANAEROBIC BACTERIOLOGY FOR THE CLINICAL LABORATORY by James I. Mangels, MA, CLS, MT(ASCP) Consultant Microbiology Consulting Services Santa Rosa, CA Course Number: DL-974 3.0 CE/Contact Hour Level of Difficulty: Intermediate © California Association for Medical Laboratory Technology. Permission to reprint any part of these materials, other than for credit from CAMLT, must be obtained in writing from the CAMLT Executive Office. CAMLT is approved by the California Department of Health Services as a CA CLS Accrediting Agency (#0021) and this course is is approved by ASCLS for the P.A.C.E. ® Program (#519) 1895 Mowry Ave, Suite 112 Fremont, CA 94538-1766 Phone: 510-792-4441 FAX: 510-792-3045 Notification of Distance Learning Deadline All continuing education units required to renew your license must be earned no later than the expiration date printed on your license. If some of your units are made up of Distance Learning courses, please allow yourself enough time to retake the test in the event you do not pass on the first attempt. CAMLT urges you to earn your CE units early!. CAMLT Distance Learning Course # DL-974 1 © California Association for Medical Laboratory Technology Outline A. Introduction B. What are anaerobic bacteria? Concepts of anaerobic bacteriology C. Why do we need to identify anaerobes? D. Normal indigenous anaerobic flora; the incidence of anaerobes at various body sites E. Anaerobic infections; most common anaerobic infections F. Specimen collection and transport; acceptance and rejection criteria G. Processing of clinical specimens 1. Microscopic examination 2. -
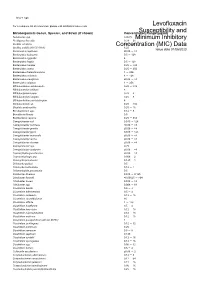
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

(12) United States Patent (10) Patent No.: US 8,501,463 B2 Cox Et Al
USOO85O1463B2 (12) United States Patent (10) Patent No.: US 8,501,463 B2 Cox et al. (45) Date of Patent: Aug. 6, 2013 (54) ANAEROBC PRODUCTION OF HYDROGEN (56) References Cited AND OTHER CHEMICAL PRODUCTS U.S. PATENT DOCUMENTS (75) Inventors: Marion E. Cox, Morgan Hill, CA (US); 5,350,685 A 9/1994 Taguchi et al. Laura M. Nondorf, Morgan Hill, CA 5,464,539 A 11/1995 Ueno et al. 6,090,266 A 7/2000 Roychowdhury (US); Steven M. Cox, Morgan Hill, CA 6,251,643 B1 6/2001 Hansen et al. (US) 6,299,774 B1 * 10/2001 Ainsworth et al. ........... 210,603 6,342,378 B1 1/2002 Zhang et al. (73) Assignee: Anaerobe Systems, Morgan Hill, CA 6,569,332 B2 * 5/2003 Ainsworth et al. ........... 210,603 2004/0050778 A1 3/2004 Noike et al. (US) 2004/O115782 A1 6/2004 Paterek (*) Notice: Subject to any disclaimer, the term of this FOREIGN PATENT DOCUMENTS patent is extended or adjusted under 35 WO WO-2006-119052 A2 11/2006 U.S.C. 154(b) by 1347 days. OTHER PUBLICATIONS (21) Appl. No.: 11/912,881 Liu et al., 2004. Effects of Culture and Medium Conditions on Hydro gen Production from Starch Using Anaerobic Bacteria. Journal of (22) PCT Fled: Apr. 27, 2006 Bioscience and Bioengineering, vol. 98, No. 4, pp. 251-256.* Zhang et al., Distributed Computer Control of Penicillin Fermenta (86) PCT NO.: PCT/US2OO6/O16332 tion Industrial Production. Proceedings of the IEEE International Conference on Industrial Technology, 1996, pp. 52-56.* S371 (c)(1), New Brunswick, an eppenforf Company, pp. -

Investigation of Infectious Agents Associated with Arthritis by Reverse Transcription PCR of Bacterial Rrna Charles J Cox1, Karen E Kempsell2 and J S Hill Gaston1
Available online http://arthritis-research.com/content/5/1/R1 Research article Open Access Investigation of infectious agents associated with arthritis by reverse transcription PCR of bacterial rRNA Charles J Cox1, Karen E Kempsell2 and J S Hill Gaston1 1Department of Rheumatology, University of Cambridge, Cambridge 2GlaxoSmithKline Medicines Research Centre, Stevenage, UK Corresponding author: J S Hill Gaston (e-mail: [email protected]) Received: 18 July 2002 Revisions received: 11 September 2002 Accepted: 13 September 2002 Published: 11 October 2002 Arthritis Res Ther 2003, 5:R1-R8 (DOI 10.1186/ar602) © 2003 Cox et al., licensee BioMed Central Ltd (Print ISSN 1478-6354; Online ISSN 1478-6362). This is an Open Access article: verbatim copying and redistribution of this article are permitted in all media for any non-commercial purpose, provided this notice is preserved along with the article's original URL. Abstract In reactive and postinfectious arthritis the joints are generally sequencing the products obtained using universal primers, but sterile but the presence of bacterial antigens and nucleic acids could in some cases be shown to be present by amplifying with has been reported. To investigate whether organisms traffic to species specific primers. This was the case for Yersinia affected joints in these conditions, we performed reverse pseudotuberculosis and Chlamydia trachomatis. However, in transcription PCR using universal primers to amplify any arthritis thought to be related to Campylobacter infection the bacterial 16S rRNA sequences present in synovial fluid. sequences obtained were not from Campylobacter jejuni or Bacterial sequences were detected in most cases, even after C. coli, but from other Campylobacter spp. -

Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Norfloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Bacillus spp. (MTCC 297) ≤1 Minimum Inhibitory Bacillus subtilis 0.049 - 10 Bacteroides bivius Concentration≥1.56 (MIC) Data Bacteroides distasonis ≥12.5 Issue date 01/06/2020 Bacteroides fragilis 16 - 25 Bacteroides intermedius 3.13 Bacteroides oralis 3.13 Bacteroides oris (ATCC 33573) 3.13 Bacteroides ovatus ≥100 Bacteroides thetaiotaomicron ≥200 Bacteroides uniformis ≥50 Bacteroides ureolyticus ≥0.2 Bacteroides vulgatus ≥100 Bifidobacterium adolescentis ≤0.025 Bordetella bronchiseptica 1 - 8 Borrelia burgdorferi S.L. 1 - 16 Burkholderia mallei 2 - 4 Capnocytophaga ochracea ≤0.025 Chlamydia pneumonia ≥16 Chlamydia psittaci ≥16 Chlamydia trachomatis 25 Citrobacter freundii 0.39 - 1 Clostridium difficile ≥50 Clostridium perfringens ≥3.13 Clostridium septicum ≥0.78 Clostridium spiroforme 8 - 64 Corynebacterium <1 Corynebacterium diphtheriae ≥0.39 Enterobacter aerogenes 0.2 - 256 Enterobacter cloacae 0.125 - >256 Enterococcus faecalis 1.56 - 66 Escherichia coli 0.016 - >128 Eubacterium lentum ≥6.25 Fusobacterium mortiferum ≥6.25 Fusobacterium varium ≥50 Hafnia alvei ≥0.1 Helicobacter pylori 0.05 - 25 Klebsiella pneumonia 0.049 - 43 Klebsiella spp. ≤1 Lactobacillus acidophilus 500 - >1000 Lactobacillus gasseri >1000 Lactobacillus johnsonii ≥750 Lactobacillus paracasei ≥1000 Lactobacillus reuteri ≥1000 Lactobacillus salivarius ≥250 Micrococcus luteus ≥12.5 Morganella morganii ≥0.2 Mycobacterium africanum ≥2 Mycobacterium bovis ≥4 Mycobacterium tuberculosis 2 - 31.2 Neisseria meningitidis ≥0.025 Peptostreptococcus anaerobius ≥6.25 Peptostreptococcus asaccharolyticus ≥6.25 Peptostreptococcus magnus ≥0.78 Peptostreptococcus prevotii ≥6.25 Proteus mirabilis ≥0.1 Proteus spp. -

Fatal Bacteremia Caused by Campylobacter Gracilis, United States
LETTERS nosocomial transmission during that period (9). However, 9. Anderson LF, Tamne S, Brown T, Watson JP, Mullarkey C, the emergence of MDR TB in regions of high HIV prev- Zenner D, et al. Transmission of multidrug-resistant tuberculosis in the UK: a cross-sectional molecular and epidemiological study of alence is relatively recent (10), and the cases described clustering and contact tracing. Lancet Infect Dis. 2014;14:406–15. here suggest that increased vigilance for TB and MDR TB http://dx.doi.org/10.1016/S1473-3099(14)70022-2 among migrating health care workers might be required. 10. Abdool Karim SS, Churchyard GJ, Karim QA, Lawn SD. HIV infection and tuberculosis in South Africa: an urgent need to escalate the public health response. Lancet. 2009;374:921–33. Acknowledgments http://dx.doi.org/10.1016/S0140-6736(09)60916-8 We thank the families of patients who gave permission for this study and all those involved in data collection. We also thank Address for correspondence: Graham S. Cooke, Imperial College, Tim Brown, Vladyslav Nikolayevskyy, and Madeline Stone for Jefferiss Laboratories, St. Mary’s Campus, Praed St, London W21NY, the VNTR analysis and helpful comments and the staff of Public UK; email: [email protected] Health England National Mycobacterium Reference Laboratory for their assistance. This study was supported in part by the Imperial College NHS Fatal Bacteremia Caused by Trust Biomedical Research Centre, National Institute for Health Campylobacter gracilis, Research Health Protection Research Units (NIHR HPRU) in Healthcare Associated Infection and Antimicrobial Resistance United States and the NIHR HPRU in Respiratory Infections, both at Impe- rial College London in partnership with Public Health England. -

Bacteroides Gracilis, an Important Anaerobic Bacterial Pathogen CAROLINE C
JOURNAL OF CLINICAL MICROBIOLOGY, Nov. 1985, p. 799-802 Vol. 22, No. 5 0095-1137/85/110799-04$02.00/0 Copyright (C 1985, American Society for Microbiology Bacteroides gracilis, an Important Anaerobic Bacterial Pathogen CAROLINE C. JOHNSON,'* JOHN F. REINHARDT,' MARTHA A. C. EDELSTEIN,"2 MAURY E. MULLIGAN,"3 W. LANCE GEORGE,'13'4 AND SYDNEY M. FINEGOLD" 2'3'5 Medical,1 Research,2 and Pathology4 Services, Infectious Disease Section, Veterans Administration Wadsworth Medical Center, Los Angeles, California 90073, and Departments of Medicine3 and Microbiology and Immunology,5 University of California Los Angeles School of Medicine, Los Angeles, California 90024 Received 31 May 1985/Accepted 31 July 1985 Clinical isolates of agar-pitting, formate-fumarate-requiring, anaerobic gram-negative bacilli were recharacterized in consideration of the species descriptions of Bacteroides ureolyticus and the newly described B. gracilis, Campylobacter concisus, and Wolinella sp. During an 11-year period, 7.5% (101 of 1,341) of all specimens yielding anaerobes were found to contain an organism in this group. Of the 71 isolates that were available for study, 43 were B. ureolyticus, 23 were B. gracilis, and 5 were in the Wolinella-C. concisus group. The role in infection and patterns of antimicrobial susceptibility for B. ureolyticus and B. gracilis were studied. Review of the clinical data indicated that 83% of B. gracilis strains were recovered from patients with serious visceral or head and neck infection, whereas 73% of B. ureolyticus isolates were recovered from superficial soft-tissue or bone infections. The strains of B. ureolyticus were uniformly susceptible to the tested antimicrobial agents. -

Meropenem Meropenem Was Administered to Dams from Gestation Day 17 Until Lactation Day 21 at 83 to 140) for the 1 Gram Dose
HIGHLIGHTS OF PRESCRIBING INFORMATION Pediatric patients less than 3 months of age 2.3 Use in Pediatric Patients Pediatric Patients 3 Months of Age and Older Close adherence to the recommended dosage regimens is urged, especially in patients Complicated Skin and Skin Structure Infections · For pediatric patients 3 months of age and older, the Meropenem for injection (I.V.) dose with known factors that predispose to convulsive activity. Continue anti-convulsant In a study of complicated skin and skin structure infections, the adverse reactions were These highlights do not include all the information needed to use Meropenem for Recommended Meropenem for injection (I.V.) Dosage Schedule for Pediatric Patients is 10 mg/kg, 20 mg/kg or 40 mg/kg every 8 hours (maximum dose is 2 grams every therapy in patients with known seizure disorders. If focal tremors, myoclonus, or seizures similar to those listed above. The most common adverse events occurring in greater than injection (I.V.) safely and effectively. See full prescribing information for Meropenem Less than 3 Months of Age with Complicated Intra-Abdominal Infections and Normal 8 hours), depending on the type of infection (cSSSI, cIAI, intra-abdominal infection or occur, evaluate neurologically, placed on anti-convulsant therapy if not already instituted, 5% of the patients were: headache (7.8%), nausea (7.8%), constipation (7%), diarrhea for injection (I.V.) Renal Function (2.3) meningitis). See dosing table 2 below. and re-examine the dosage of Meropenem for injection (I.V.) to determine whether it (7%), anemia (5.5%), and pain (5.1%). Adverse events with an incidence of greater than · For pediatric patients weighing over 50 kg administer Meropenem for injection (I.V.) at should be decreased or discontinued. -

Bacteroides Ureolyticus, a New Species to Accommodate Strains Previously Identified As “Bacteroides Corrodens, Anaerobic”
INTERNATIONAL JOURNALOF SYSTEMATIC BACTERIOLOGY, Apr. 1978, p. 197-200 Vol. 28, No. 2 0020-7713/78/0028-0197$02.00/0 Copyright 0 1978 International Association of Microbiological Societies Printed in U.S. A. Bacteroides ureolyticus, a New Species to Accommodate Strains Previously Identified as “Bacteroides corrodens, Anaerobic” FRANCIS L. JACKSON AND YVONNE E. GOODMAN Department of Medical Bacteriology, University of Alberta, Edmonton, Alberta, Canada The name Bacteroides ureolyticus is proposed for a species to accommodate strains of gram-negative, urease-positive, anaerobic, corroding rods previously incorrectly referred to as Bacteroides corrodens. The organisms are catalase negative and reduce nitrate. In peptone-yeast-glucose medium, growth is en- hanced by the addition of fumarate and formate, and succinate is the major end product. Conventional carbohydrate fermentation tests are negative. The oxidase test is positive and is markedly inhibited by azide. Absorption bands of reduced cytochromes are seen by visual spectroscopy at 550 nm (cytochrome c) and 555-560 nm (b-type cytochrome); no cytochrome a band was detected. Most strains hydrolyze gelatin and casein. Strains with very weak proteolytic activity are also encountered. The cells are nonflagellated but may show “twitching motility.” Electron micrographs of five of seven strains reveal polar pili. Strains that lack pili do not produce spreading colonies or show twitching motility. The guanine-plus-cytosine content of the deoxyribonucleic acid is in the range of 28.0 to 30.0 mol%. Strain NCTC 10941 is designated as the type strain. Eiken (2) applied the name Bacteroides cor- MATERIALS AND METHODS rodens to certain gram-negative rods that Bacterial strains. -

Original Article the Human Oral Microbiome Database: a Web Accessible Resource for Investigating Oral Microbe Taxonomic and Genomic Information
Database, Vol. 2010, Article ID baq013, doi:10.1093/database/baq013 ............................................................................................................................................................................................................................................................................................. Original article The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information Tsute Chen1,2,*, Wen-Han Yu1, Jacques Izard1,2, Oxana V. Baranova1, Abirami Lakshmanan1 and Floyd E. Dewhirst1,2 1The Forsyth Institute, Boston, MA 02115, USA and 2Department of Oral Medicine, Infection and Immunity, Harvard School of Dental Medicine, Boston, MA 02115, USA *Corresponding author: Tel: +1 617 892 8359; Fax: +1 617 262 5200; Email: [email protected] Submitted 25 January 2010; Revised 28 May 2010; Accepted 20 June 2010 ............................................................................................................................................................................................................................................................................................. The human oral microbiome is the most studied human microflora, but 53% of the species have not yet been validly named and 35% remain uncultivated. The uncultivated taxa are known primarily from 16S rRNA sequence information. Sequence information tied solely to obscure isolate or clone numbers, and usually lacking accurate phylogenetic placement,