Updating the Genomic Taxonomy and Epidemiology of Campylobacter Hyointestinalis Received: 3 October 2017 David A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies on the Detection Methods of Campylobacter and Faecal Indicator Bacteria in Drinking Water
Tarja Pitkänen Tarja Pitkänen Tarja Studies on the Detection Tarja Pitkänen Methods of Campylobacter RESEARCH Studies on the Detection Methods of RESEARCH Campylobacter and Faecal Indicator Bacteria and Faecal Indicator in Drinking Water Bacteria in Drinking Water Indicator Bacteria in Drinking Water Drinking in Bacteria Indicator Methods Detection the on Studies Faecal contamination of drinking water and subsequent waterborne gastrointestinal infection outbreaks are a major public health concern. In this study, faecal indicator bacteria were detected in 10% of the groundwater samples analysed. The main on-site hazards to water safety at small community water supplies included inadequate well construction and maintenance, an insufficient depth of the protective soil layer and bank filtration. As a preventive measure, the upgrading of the water treatment processes and utilization of disinfection at small Finnish groundwater supplies are recommended. More efficient and specific and less time-consuming methods for enumeration and typing of E. coli and coliform bacteria from non-disinfected water as well as for cultivation and molecular detection and typing of Campylobacter were found in the study. These improvements in methodology for the analysis of the faecal bacteria from water might promote public health protection as they Campylobacter could be anticipated to result in very important time savings and improve the tracking of faecal contamination source in waterborne outbreak investigations. and Faecal Faecal and .!7BC5<2"HIGEML! National Institute for Health and Welfare P.O. Box 30 (Mannerheimintie 166) FI-00271 Helsinki, Finland Telephone: +358 20 610 6000 39 ISBN 978-952-245-319-8 39 2010 39 www.thl.fi Tarja Pitkänen Studies on the Detection Methods of Campylobacter and Faecal Indicator Bacteria in Drinking Water ACADEMIC DISSERTATION To be presented with the permission of the Faculty of Science and Forestry of the University of Eastern Finland for public examination in auditorium, MediTeknia Building, on October 1st, 2010 at 12 o’clock noon. -

Campylobacteriosis: a Global Threat
ISSN: 2574-1241 Volume 5- Issue 4: 2018 DOI: 10.26717/BJSTR.2018.11.002165 Muhammad Hanif Mughal. Biomed J Sci & Tech Res Review Article Open Access Campylobacteriosis: A Global Threat Muhammad Hanif Mughal* Homeopathic Clinic, Rawalpindi, Islamabad, Pakistan Received: : November 30, 2018; Published: : December 10, 2018 *Corresponding author: Muhammad Hanif Mughal, Homeopathic Clinic, Rawalpindi-Islamabad, Pakistan Abstract Campylobacter species account for most cases of human gastrointestinal infections worldwide. In humans, Campylobacter bacteria cause illness called campylobacteriosis. It is a common problem in the developing and industrialized world in human population. Campylobacter species extensive research in many developed countries yielded over 7500 peer reviewed articles. In humans, most frequently isolated species had been Campylobacter jejuni, followed by Campylobactercoli Campylobacterlari, and lastly Campylobacter fetus. C. jejuni colonizes important food animals besides chicken, which also includes cattle. The spread of the disease is allied to a wide range of livestock which include sheep, pigs, birds and turkeys. The organism (5-18.6 has% of been all Campylobacter responsible for cases) diarrhoea, in an estimated 400 - 500 million people globally each year. The most important Campylobacter species associated with human infections are C. jejuni, C. coli, C. lari and C. upsaliensis. Campylobacter colonize the lower intestinal tract, including the jejunum, ileum, and colon. The main sources of these microorganisms have been traced in unpasteurized milk, contaminated drinking water, raw or uncooked meat; especially poultry meat and contact with animals. Keywords: Campylobacteriosis; Gasteritis; Campylobacter jejuni; Developing countries; Emerging infections; Climate change Introduction of which C. jejuni and 12 species of C. coli have been associated with Campylobacter cause an illness known as campylobacteriosis is a common infectious problem of the developing and industrialized world. -

A Campylobacter Integrative and Conjugative Element with a CRISPR-Cas9
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446523; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. van Vliet et al. CRISPR-Cas on Campylobacter mobile elements A Campylobacter integrative and conjugative element with a CRISPR-Cas9 system targeting competing plasmids: a history of plasmid warfare? Arnoud H.M. van Vliet 1,*, Oliver Charity 2, Mark Reuter 2 1. School of Veterinary Medicine, Department of Pathology and Infectious Diseases, University of Surrey, Guildford, United Kingdom. 2. Quadram Institute Bioscience, Microbes in the Food Chain programme, Norwich, United Kingdom. * Corresponding author. Mailing address: School of Veterinary Medicine, University of Surrey, Daphne Jackson Road, Guildford GU2 7AL, United Kingdom. Phone +44-1483-684406, E-mail: [email protected] Running title: CRISPR-Cas on Campylobacter mobile elements Keywords: Campylobacter, mobile genetic elements, plasmids, CRISPR-Cas 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446523; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. van Vliet et al. CRISPR-Cas on Campylobacter mobile elements 1 ABSTRACT 2 Microbial genomes are highly adaptable, with mobile genetic elements (MGEs) such as 3 integrative conjugative elements (ICE) mediating the dissemination of new genetic information 4 throughout bacterial populations. -

Potential Role of Microbiome in Oncogenesis, Outcome Prediction
Oral Oncology 99 (2019) 104453 Contents lists available at ScienceDirect Oral Oncology journal homepage: www.elsevier.com/locate/oraloncology Review Potential role of microbiome in oncogenesis, outcome prediction and therapeutic targeting for head and neck cancer T ⁎ Ester Orlandia,b, , Nicola Alessandro Iacovellib, Vincenzo Tombolinic, Tiziana Rancatid, Antonella Polimenie, Loris De Ceccof, Riccardo Valdagnia,d,g, Francesca De Felicec a Department of Radiotherapy 1, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy b Department of Radiotherapy 2, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy c Department of Radiotherapy, Policlinico Umberto I, “Sapienza” University of Rome, Rome, Italy d Prostate Cancer Program, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy e Department of Oral and Maxillo Facial Sciences, Policlinico Umberto I, “Sapienza” University of Rome, Italy f Integrated Biology Platform, Department of Applied Research and Technology Development, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Milan, Italy g Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy ARTICLE INFO ABSTRACT Keywords: In the last decade, human microbiome research is rapidly growing involving several fields of clinical medicine Head and neck cancer and population health. Although the microbiome seems to be linked to all sorts of diseases, cancer has the Biomarkers biggest potential to be investigated. Microbiome Following the publication of the National Institute of Health - Human Microbiome Project (NIH-HMP), the Microbiota link between Head and Neck Cancer (HNC) and microbiome seems to be a fast-moving field in research area. Oncogenesis However, robust evidence-based literature is still quite scarce. Nevertheless the relationship between oral mi- Radiotherapy Immunotherapy crobiome and HNC could have important consequences for prevention and early detection of this type of tumors. -

3. Jedna Z Krvných Skupín Systému AB0; 4. Skr. Bukálny. B – Symbo
B – 1. symbol pre bel; 2. chem. značka prvku →bór; 3. jedna z krvných skupín systému AB0; 4. skr. bukálny. B – symbol pre hustotu magnetického toku. B. – skr. pre →Bacillus. B-ALP – skr. angl. bone alcalic phosphatase kostná alkalická fosfatáza. B-bunky – 1. syn. -bunky Langerhansových ostrovčekov; →pankreas; 2. syn. B-lymfocyty; →lymfocyty. B-komplex – multivitamínový prípravok vitamínov B. B-lymfocyty – [B podľa Fabriciovej burzy, imunol. orgánu vtákov] B bunky, druh lymfocytov, kt. sa zúčastňuje na humorálnej imunite (tvorbe protilátok) a niekt. ďalších imunitných funkciách. Povrchové molekuly sú CD 19,20. Receptorom pre antigén je membránový imunoglobulín. B-reťazec – jeden z polypeptidových reťazcov →inzulínu. B-vírus – 1. vírus →hepatitídy B.; 2. vírus zo skupiny Cercopithecine herpes virus 1. B-vlákna →nerv. b – skr. 1. pre barn; 2. skr. angl. born narodený; 3. skr. báza (genet. označenie dĺţky sekvencie nukleotidov, napr. 50 b = sekvencia 50 nukleotidov). b-vlna – pozit. kmit s vyskou amplitúdou v →elektroretinograme nasledujúci po vlne a, prejav komplexnej aktivity vrstvy bipolárnych buniek. – - – predpona označujúca 1. staršie označenie druhého atómu uhlíka v reťazci, na kt. sa pripája hlavná funkčná skupina, napr. kys. -hydroxymaslová správ. kys. 3-hydroxymaslová; 2. špecifická rotácia opticky aktívnej látky, napr. -D-glukóza; 3. orientácia exocyklického atómu al. skupiny, napr. cholest-5-en-3--ol; 4. plazmatický proteín, kt. migruje v elektroforéze v pruhu - lipoproteín; 5. člen série príbuzných chem. látok, najmä série stereoizomérov, izomérov, polymérov al. alotroických foriem, napr. -karotén; 6. -lúč. B2 – staršie označenie pre CD21. B4 – staršie označenie pre CD 19. B19 virus – ľudský parvovírus z čeľade Parvoviridae, značne rozšírený, vyvolávajúci obvykle inaparentné infekcie. -

974-Form.Pdf
California Association for Medical Laboratory Technology Distance Learning Program ANAEROBIC BACTERIOLOGY FOR THE CLINICAL LABORATORY by James I. Mangels, MA, CLS, MT(ASCP) Consultant Microbiology Consulting Services Santa Rosa, CA Course Number: DL-974 3.0 CE/Contact Hour Level of Difficulty: Intermediate © California Association for Medical Laboratory Technology. Permission to reprint any part of these materials, other than for credit from CAMLT, must be obtained in writing from the CAMLT Executive Office. CAMLT is approved by the California Department of Health Services as a CA CLS Accrediting Agency (#0021) and this course is is approved by ASCLS for the P.A.C.E. ® Program (#519) 1895 Mowry Ave, Suite 112 Fremont, CA 94538-1766 Phone: 510-792-4441 FAX: 510-792-3045 Notification of Distance Learning Deadline All continuing education units required to renew your license must be earned no later than the expiration date printed on your license. If some of your units are made up of Distance Learning courses, please allow yourself enough time to retake the test in the event you do not pass on the first attempt. CAMLT urges you to earn your CE units early!. CAMLT Distance Learning Course # DL-974 1 © California Association for Medical Laboratory Technology Outline A. Introduction B. What are anaerobic bacteria? Concepts of anaerobic bacteriology C. Why do we need to identify anaerobes? D. Normal indigenous anaerobic flora; the incidence of anaerobes at various body sites E. Anaerobic infections; most common anaerobic infections F. Specimen collection and transport; acceptance and rejection criteria G. Processing of clinical specimens 1. Microscopic examination 2. -

The Global View of Campylobacteriosis
FOOD SAFETY THE GLOBAL VIEW OF CAMPYLOBACTERIOSIS REPORT OF AN EXPERT CONSULTATION UTRECHT, NETHERLANDS, 9-11 JULY 2012 THE GLOBAL VIEW OF CAMPYLOBACTERIOSIS IN COLLABORATION WITH Food and Agriculture of the United Nations THE GLOBAL VIEW OF CAMPYLOBACTERIOSIS REPORT OF EXPERT CONSULTATION UTRECHT, NETHERLANDS, 9-11 JULY 2012 IN COLLABORATION WITH Food and Agriculture of the United Nations The global view of campylobacteriosis: report of an expert consultation, Utrecht, Netherlands, 9-11 July 2012. 1. Campylobacter. 2. Campylobacter infections – epidemiology. 3. Campylobacter infections – prevention and control. 4. Cost of illness I.World Health Organization. II.Food and Agriculture Organization of the United Nations. III.World Organisation for Animal Health. ISBN 978 92 4 156460 1 _____________________________________________________ (NLM classification: WF 220) © World Health Organization 2013 All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications –whether for sale or for non-commercial distribution– should be addressed to WHO Press through the WHO web site (www.who.int/about/licensing/copyright_form/en/index. html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Oral and Fecal Campylobacter Concisus Strains Perturb Barrier Function by Apoptosis Induction in HT-29/B6 Intestinal Epithelial Cells
Oral and Fecal Campylobacter concisus Strains Perturb Barrier Function by Apoptosis Induction in HT-29/B6 Intestinal Epithelial Cells Hans Linde Nielsen1, Henrik Nielsen1, Tove Ejlertsen2, Jørgen Engberg3, Dorothee Gu¨ nzel4, Martin Zeitz5, Nina A. Hering5, Michael Fromm4,Jo¨ rg-Dieter Schulzke5*, Roland Bu¨ cker5 1 Department of Infectious Diseases, Aalborg Hospital, Aarhus University Hospital, Aalborg, Denmark, 2 Department of Clinical Microbiology, Aalborg Hospital, Aarhus University Hospital, Aalborg, Denmark, 3 Department of Clinical Microbiology, Slagelse Hospital, Slagelse, Denmark, 4 Institute of Clinical Physiology, Charite´ Universita¨tsmedizin Berlin, Berlin, Germany, 5 Department of Gastroenterology, Infectious Diseases and Rheumatology, Division of General Medicine and Nutrition, Charite´ Universita¨tsmedizin Berlin, Berlin, Germany Abstract Campylobacter concisus infections of the gastrointestinal tract can be accompanied by diarrhea and inflammation, whereas colonization of the human oral cavity might have a commensal nature. We focus on the pathophysiology of C. concisus and the effects of different clinical oral and fecal C. concisus strains on human HT-29/B6 colon cells. Six oral and eight fecal strains of C. concisus were isolated. Mucus-producing HT-29/B6 epithelial monolayers were infected with the C. concisus strains. Transepithelial electrical resistance (Rt) and tracer fluxes of different molecule size were measured in Ussing chambers. Tight junction (TJ) protein expression was determined by Western blotting, and subcellular TJ distribution was analyzed by confocal laser-scanning microscopy. Apoptosis induction was examined by TUNEL-staining and Western blot of caspase-3 activation. All strains invaded confluent HT-29/B6 cells and impaired epithelial barrier function, characterized by a time- and dose-dependent decrease in Rt either after infection from the apical side but even more from the basolateral compartment. -
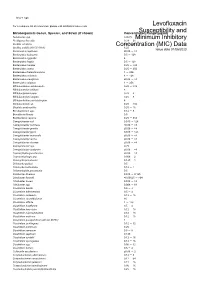
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

Review Campylobacter As a Major Foodborne Pathogen
REVIEW CAMPYLOBACTER AS A MAJOR FOODBORNE PATHOGEN: A REVIEW OF ITS CHARACTERISTICS, PATHOGENESIS, ANTIMICROBIAL RESISTANCE AND CONTROL Ahmed M. Ammar1, El-Sayed Y. El-Naenaeey1, Marwa I. Abd El-Hamid1, Attia A. El-Gedawy2 and Rania M. S. El- Malt*3 Address: Rania Mohamed Saied El-Malt 1 Zagazig University, Faculty of Veterinary Medicine, Department of Microbiology, 19th Saleh Abo Rahil Street, El-Nahal, 44519, Zagazig, Sharkia, Egypt 2 Animal health Research Institute, Department of Bacteriology, Tuberculosis unit, Nadi El-Seid Street,12618 Dokki, Giza, Egypt 3 Animal health Research Institute, Department of Microbiology, El-Mohafza Street, 44516, Zagazig, Sharkia, Egypt, +201061463064 *Corresponding author: [email protected] ABSTRACT Campylobacter, mainly Campylobacter jejuni is viewed as one of the most well-known reasons of foodborne bacterial diarrheal sickness in people around the globe. The genus Campylobacter contains 39 species (spp.) and 16 sub spp. Campylobacter is microaerophilic, Gram negative, spiral- shaped rod with characteristic cork screw motility. It is colonizing the digestive system of numerous wild and household animals and birds, particularly chickens. Intestinal colonization brings about transporter/carrier healthy animals. Consequently, the utilization of contaminated meat, especially chicken meat is the primary source of campylobacteriosis in humans and chickens are responsible for an expected 80% of human campylobacter infection. Interestingly, in contrast with the most recent published reviews that cover specific aspects of campylobacter/campylobacteriosis, this review targets the taxonomy, biological characteristics, identification and habitat of Campylobacter spp. Moreover, it discusses the pathogenesis, resistance to antimicrobial agents and public health significance of Campylobacter spp. Finally, it focuses on the phytochemicals as intervention strategies used to reduce Campylobacter spp.in poultry production. -

(12) United States Patent (10) Patent No.: US 8,501,463 B2 Cox Et Al
USOO85O1463B2 (12) United States Patent (10) Patent No.: US 8,501,463 B2 Cox et al. (45) Date of Patent: Aug. 6, 2013 (54) ANAEROBC PRODUCTION OF HYDROGEN (56) References Cited AND OTHER CHEMICAL PRODUCTS U.S. PATENT DOCUMENTS (75) Inventors: Marion E. Cox, Morgan Hill, CA (US); 5,350,685 A 9/1994 Taguchi et al. Laura M. Nondorf, Morgan Hill, CA 5,464,539 A 11/1995 Ueno et al. 6,090,266 A 7/2000 Roychowdhury (US); Steven M. Cox, Morgan Hill, CA 6,251,643 B1 6/2001 Hansen et al. (US) 6,299,774 B1 * 10/2001 Ainsworth et al. ........... 210,603 6,342,378 B1 1/2002 Zhang et al. (73) Assignee: Anaerobe Systems, Morgan Hill, CA 6,569,332 B2 * 5/2003 Ainsworth et al. ........... 210,603 2004/0050778 A1 3/2004 Noike et al. (US) 2004/O115782 A1 6/2004 Paterek (*) Notice: Subject to any disclaimer, the term of this FOREIGN PATENT DOCUMENTS patent is extended or adjusted under 35 WO WO-2006-119052 A2 11/2006 U.S.C. 154(b) by 1347 days. OTHER PUBLICATIONS (21) Appl. No.: 11/912,881 Liu et al., 2004. Effects of Culture and Medium Conditions on Hydro gen Production from Starch Using Anaerobic Bacteria. Journal of (22) PCT Fled: Apr. 27, 2006 Bioscience and Bioengineering, vol. 98, No. 4, pp. 251-256.* Zhang et al., Distributed Computer Control of Penicillin Fermenta (86) PCT NO.: PCT/US2OO6/O16332 tion Industrial Production. Proceedings of the IEEE International Conference on Industrial Technology, 1996, pp. 52-56.* S371 (c)(1), New Brunswick, an eppenforf Company, pp. -

MICRO-ORGANISMS and RUMINANT DIGESTION: STATE of KNOWLEDGE, TRENDS and FUTURE PROSPECTS Chris Mcsweeney1 and Rod Mackie2
BACKGROUND STUDY PAPER NO. 61 September 2012 E Organización Food and Organisation des Продовольственная и cельскохозяйственная de las Agriculture Nations Unies Naciones Unidas Organization pour организация para la of the l'alimentation Объединенных Alimentación y la United Nations et l'agriculture Наций Agricultura COMMISSION ON GENETIC RESOURCES FOR FOOD AND AGRICULTURE MICRO-ORGANISMS AND RUMINANT DIGESTION: STATE OF KNOWLEDGE, TRENDS AND FUTURE PROSPECTS Chris McSweeney1 and Rod Mackie2 The content of this document is entirely the responsibility of the authors, and does not necessarily represent the views of the FAO or its Members. 1 Commonwealth Scientific and Industrial Research Organisation, Livestock Industries, 306 Carmody Road, St Lucia Qld 4067, Australia. 2 University of Illinois, Urbana, Illinois, United States of America. This document is printed in limited numbers to minimize the environmental impact of FAO's processes and contribute to climate neutrality. Delegates and observers are kindly requested to bring their copies to meetings and to avoid asking for additional copies. Most FAO meeting documents are available on the Internet at www.fao.org ME992 BACKGROUND STUDY PAPER NO.61 2 Table of Contents Pages I EXECUTIVE SUMMARY .............................................................................................. 5 II INTRODUCTION ............................................................................................................ 7 Scope of the Study ...........................................................................................................