Targeting Microcephaly Kinases in Brain Tumors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -
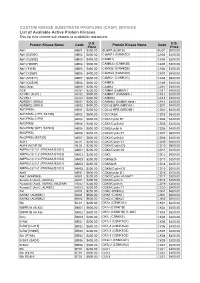
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This List May Change with Respect to Availability and Pricing
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This list may change with respect to availability and pricing. Protein Kinase Name Code U.S. Protein Kinase Name Code U.S. Price Price Abl1 AB01 $200.00 BUBR1(BUB1B) BU01 $600.00 Abl1 [E255K] AB02 $400.00 CaMK1δ (CAMK1D) CA03 $200.00 Abl1 [G250E] AB03 $400.00 CAMK1γ CA04 $200.00 Abl1 [H369P] AB04 $600.00 CAMK2α (CAMK2B) CA05 $200.00 Abl1 [T315I] AB05 $400.00 CaMK2β (CAMK2B) CA06 $200.00 Abl1 [Y253F] AB06 $400.00 CaMK2δ (CAMK2D) CA07 $400.00 Abl1 [M351T] AB07 $600.00 CaMK2γ (CAMK2G) CA08 $600.00 Abl1 [Q252H] AB08 $600.00 CAMK3γ CA09 $200.00 Abl2 (Arg) AB09 $200.00 CAMK4 CA10 $200.00 ACK AC01 $200.00 CAMK4 (CaMKIV ) CA11 $400.00 ACVR1 (ALK2 ) AC02 $400.00 CAMKK1 (CAMKKA ) CA12 $200.00 ACVRL1 AC03 $400.00 CAMKK2 CA13 $200.00 ADRBK1 (GRK2) AD01 $200.00 CAMKK2 (CaMKK beta ) CA14 $400.00 ADRBK2 (GRK3) AD02 $400.00 CDC42 BPA (MRCKA ) CD01 $400.00 Akt1/PKBα AK01 $200.00 CDC42 BPB (MRCKB ) CD02 $400.00 Akt1/PKBα [δPH, S473D] AK02 $600.00 CDC7/ASK CD03 $600.00 Akt1/PKBα [δPH] AK03 $600.00 CDK1/cyclin B1 CD04 $400.00 Akt2/PKBβ AK04 $200.00 CDK1/CyclinA2 CD05 $200.00 Akt2/PKBβ [δPH, S474D] AK05 $600.00 CDK2/cyclin A CD06 $400.00 Akt3/PKBγ AK06 $200.00 CDK2/Cyclin E1 CD07 $600.00 Akt3/PKBγ [S472D] AK07 $600.00 CDK2/CyclinA2 CD08 $200.00 ALK1 AL01 $200.00 CDK3/Cyclin E1 CD09 $600.00 ALK4 (ACVR1B) AL02 $200.00 CDK4//Cyclin D3 CD10 $600.00 AMPKα1/β1/γ1 (PRKAA1/B1/G1) AM01 $200.00 CDK4/Cyclin D1 CD11 $200.00 AMPKα1/β1/γ2 (PRKAA1/B1/G2) AM02 $200.00 CDK5 CD12 $600.00 -

Mouse Cdc42bpa Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Cdc42bpa Knockout Project (CRISPR/Cas9) Objective: To create a Cdc42bpa knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Cdc42bpa gene (NCBI Reference Sequence: NM_001033285 ; Ensembl: ENSMUSG00000026490 ) is located on Mouse chromosome 1. 37 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 37 (Transcript: ENSMUST00000111117). Exon 3~5 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 3 starts from about 5.22% of the coding region. Exon 3~5 covers 6.33% of the coding region. The size of effective KO region: ~8696 bp. The KO region does not have any other known gene. Page 1 of 10 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 5 37 Legends Exon of mouse Cdc42bpa Knockout region Page 2 of 10 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 3 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 5 is aligned with itself to determine if there are tandem repeats. -

Molecular Classification of Diffuse Cerebral WHO Grade II/III Gliomas
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2015 Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups Weller, Michael ; Weber, Ruthild G ; Willscher, Edith ; Riehmer, Vera ; Hentschel, Bettina ; Kreuz, Markus ; Felsberg, Jörg ; Beyer, Ulrike ; Löffler-Wirth, Henry ; Kaulich, Kerstin ; Steinbach, Joachim P ; Hartmann, Christian ; Gramatzki, D ; Schramm, J ; Westphal, M ; Schackert, G ; Simon, M ; Martens, T ; Boström, J ; Hagel, C ; Sabel, M ; Krex, D ; Tonn, J C ; Wick, W ; Noell, S ; Schlegel, U ; Radlwimmer, B ; Pietsch, T ; Loeffler, M ; von Deimling, A ; Binder, H ; Reifenberger, G Abstract: Cerebral gliomas of World Health Organization (WHO) grade II and III represent a major challenge in terms of histological classification and clinical management. Here, we asked whether large- scale genomic and transcriptomic profiling improves the definition of prognostically distinct entities. We performed microarray-based genome- and transcriptome-wide analyses of primary tumor samples from a prospective German Glioma Network cohort of 137 patients with cerebral gliomas, including 61 WHO grade II and 76 WHO grade III tumors. Integrative bioinformatic analyses were employed to define molecular subgroups, which were then related to histology, molecular biomarkers, including isocitrate de- hydrogenase 1 or 2 (IDH1/2) mutation, 1p/19q co-deletion and telomerase reverse transcriptase (TERT) promoter mutations, and patient outcome. Genomic profiling identified five distinct glioma groups, in- cluding three IDH1/2 mutant and two IDH1/2 wild-type groups. Expression profiling revealed evidence for eight transcriptionally different groups (five IDH1/2 mutant, three IDH1/2 wild type), which were only partially linked to the genomic groups. -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA. -

Anti-CDC42BPA Polyclonal Antibody (CABT- BL1018) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-CDC42BPA polyclonal antibody (CABT- BL1018) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Immunogen Peptide corresponding to the amino acids 1671-1685 of human MRCK alpha Isotype IgG Source/Host Rabbit Species Reactivity Human Conjugate Unconjugated Applications WB, ICC Buffer The reagent is provided in phosphate buffered saline (PBS) containing26mM sodium azide and 0.2% (w/v) high-grade protease free Bovine Serum Albumin (BSA) as a stabilizing agent. Preservative 26mM Sodium Azide Storage Store at 2-8°C. Do not use after expiration date stamped on vial label. For long-term storage aliquot and store at -20°C. Avoid freeze/thaw cycles. BACKGROUND Introduction The protein encoded by this gene is a member of the Serine/Threonine protein kinase family. This kinase contains multiple functional domains. Its kinase domain is highly similar to that of the myotonic dystrophy protein kinase (DMPK). This kinase also contains a Rac interactive binding (CRIB) domain, and has been shown to bind CDC42. It may function as a CDC42 downstream effector mediating CDC42 induced peripheral actin formation, and promoting cytoskeletal reorganization. Multiple alternatively spliced transcript variants have been described, and the full- length nature of two of them has been reported. [provided by RefSeq, Jul 2008] GENE INFORMATION 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Entrez Gene ID 8476 Protein Refseq NP_003598 UniProt ID Q5VT25 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 2 © Creative Diagnostics All Rights Reserved. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

Supplementary Table 5.List of the 220 Most Frequently Amplified Genes In
Supplementary Table 5. List of the 220 most frequently amplified genes in this study. The table includes their chromosomal location, the amplification frequency in ER-positive female breast cancer with associated p-value for difference in proportions, the preference for surrogate intrinsic molecular subtype, and associations with clinical, pathological and genetic characteristics. Potentially druggable gene categories, clinical actionability and known drug interactions are indicated per gene. Gene Full name chr location % amp in FFPE % amps in FF total % amp % amp ER+ FBC* p-value MBC vs ER+ FBC** % in lumA-like % in lumB-like p-value BRCA2 germline Age Hist type ER status PR status HER2 status Grade MAI Size LN SNV load PIK3CA mut KM (OS)*** KM (5Y OS)*** druggable gene category# clinically actionable?## known drug interactions?### THBS1 thrombospondin 1 15q14 37% 9% 30% 0.1% <0.0001 23% 35% 0.128 ns ns ns ns ns ns ns ns ns ns ns ns 0.642 p=0.832 cell surface, tumor suppressor, drug resistance, external side of plasma membrane no none PRKDC protein kinase, DNA-activated, catalytic polypeptide 8q11.21 35% 7% 27% 10.9% <0.0001 26% 30% 0.595 ns ns ns ns ns ns ns ns ns ns ns ns 0.838 p=0.903 (serine threonine) kinase, druggable genome, PI3 kinase, tumor suppressor, TF complex, TF binding, DNA repair yes DNA-PK INHIBITOR V (DNA-PK inhibitor); WORTMANNIN (PI3K inhibitor); SF1126 (PI3 kinase/mTOR inhibitor) TBX3 T-box 3 12q24.21 34% 7% 27% 0.1% <0.0001 20% 35% 0.053 ns ns ns ns ns ns ns ns ns ns ns ns 0.439 p=0.264 tumor suppressor, TF binding -

Evolution and Structure of Clinically Relevant Gene Fusions in Multiple Myeloma
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2020 Evolution and structure of clinically relevant gene fusions in multiple myeloma Steven M. Foltz Qingsong Gao Christopher J. Yoon Hua Sun Lijun Yao See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Authors Steven M. Foltz, Qingsong Gao, Christopher J. Yoon, Hua Sun, Lijun Yao, Yize Li, Reyka G. Jayasinghe, Song Cao, Justin King, Daniel R. Kohnen, Mark A. Fiala, Li Ding, and Ravi Vij ARTICLE https://doi.org/10.1038/s41467-020-16434-y OPEN Evolution and structure of clinically relevant gene fusions in multiple myeloma Steven M. Foltz 1,2, Qingsong Gao 1,2, Christopher J. Yoon1,2, Hua Sun1,2, Lijun Yao1,2, Yize Li1,2, ✉ ✉ Reyka G. Jayasinghe1,2, Song Cao1,2, Justin King1, Daniel R. Kohnen1, Mark A. Fiala1, Li Ding1,2,3,4 & Ravi Vij1,4 Multiple myeloma is a plasma cell blood cancer with frequent chromosomal translocations leading to gene fusions. To determine the clinical relevance of fusion events, we detect gene 1234567890():,; fusions from a cohort of 742 patients from the Multiple Myeloma Research Foundation CoMMpass Study. Patients with multiple clinic visits enable us to track tumor and fusion evolution, and cases with matching peripheral blood and bone marrow samples allow us to evaluate the concordance of fusion calls in patients with high tumor burden. We examine the joint upregulation of WHSC1 and FGFR3 in samples with t(4;14)-related fusions, and we illustrate a method for detecting fusions from single cell RNA-seq. -

Supplementary Table S1. Summary of the Six Next-Generation Sequencing (NGS) Studies Containing 241 Paired Melanoma Tumor/Normal Samples
Supplementary Table S1. Summary of the six next-generation sequencing (NGS) studies containing 241 paired melanoma tumor/normal samples Study NGS # paired tumor-normal Tumor subtype Reference ID technology samples 1 Whole genome 25 23 cutaneous, 2 acral Berger et al., 2012 (1) 95 cutaneous, 5 acral, 2 mucosal, 1 uveal, and 18 2 Whole exome 121 Hodis et al., 2012 (2) unknown 61 cutaneous, 14 acral, 7 mucosal, 5 uveal, and 12 Krauthammer et al., 2012 3 Whole exome 99* unknown (3) 4 Whole exome 7 7 cutaneous Nikolaev et al., 2012 (4) 5 Whole exome 8 8 cutaneous Stark et al., 2012 (5) 6 Whole exome 14 14 cutaneous Wei et al., 2011 (6) 187 cutaneous, 19 acral, 9 mucosal, 6 uveal, and 30 Total 241# unknown *48 tumor samples without normal samples were excluded from our study. #23 paired samples in Berger et al. (2012) were used in Hodis et al. (2012). In addition, there were 10 samples without any mutations in Krauthammer et al. (2012). These samples were excluded in our analysis. 1 Supplementary Table S2. Summary of known driver mutations detected in the 241 melanoma samples Mutation* Type # samples BRAF GNAQ GNA11 KIT NRAS present Acral 17 3 1 0 0 0 2 Mucosal 7 2 1 0 0 0 1 Uveal 6 3 0 0 3 0 0 Cutaneous 182 138 99 0 0 1 38 Unknown 29 26 20 0 0 0 6 Total 172 121 241 0 (0%) 3 (1.2%) 1 (0.4%) 47 (19.5%) (frequency) (71.3%) (50.2%) *Includes the somatic point mutations identified by the Vanderbilt melanoma SNaPshot assay and known to be functional and actionable (7). -
![CDC42 Binding Protein Kinase Alpha (CDC42BPA) Mouse Monoclonal Antibody [Clone ID: OTI7B9] Product Data](https://docslib.b-cdn.net/cover/5365/cdc42-binding-protein-kinase-alpha-cdc42bpa-mouse-monoclonal-antibody-clone-id-oti7b9-product-data-4635365.webp)
CDC42 Binding Protein Kinase Alpha (CDC42BPA) Mouse Monoclonal Antibody [Clone ID: OTI7B9] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA808223 CDC42 binding protein kinase alpha (CDC42BPA) Mouse Monoclonal Antibody [Clone ID: OTI7B9] Product data: Product Type: Primary Antibodies Clone Name: OTI7B9 Applications: WB Recommend Dilution: WB 1:2000 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 1419-1638 of human CDC42BPA(NP_055641) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Predicted Protein Size: 185.9 kDa Gene Name: CDC42 binding protein kinase alpha Database Link: NP_055641 Entrez Gene 8476 Human Background: The protein encoded by this gene is a member of the Serine/Threonine protein kinase family. This kinase contains multiple functional domains. Its kinase domain is highly similar to that of the myotonic dystrophy protein kinase (DMPK). This kinase also contains a Rac interactive binding (CRIB) domain, and has been shown to bind CDC42. It may function as a CDC42 downstream effector mediating CDC42 induced peripheral actin formation, and promoting cytoskeletal reorganization. Multiple alternatively spliced transcript variants have been described, and the full-length nature of two of them has been reported. [provided by RefSeq, Jul 2008] Synonyms: MRCK; MRCKA; PK428 Protein Families: Druggable Genome, Protein Kinase This product is to be used for laboratory only. -

Kinome Expression Profiling to Target New Therapeutic Avenues in Multiple Myeloma
Plasma Cell DIsorders SUPPLEMENTARY APPENDIX Kinome expression profiling to target new therapeutic avenues in multiple myeloma Hugues de Boussac, 1 Angélique Bruyer, 1 Michel Jourdan, 1 Anke Maes, 2 Nicolas Robert, 3 Claire Gourzones, 1 Laure Vincent, 4 Anja Seckinger, 5,6 Guillaume Cartron, 4,7,8 Dirk Hose, 5,6 Elke De Bruyne, 2 Alboukadel Kassambara, 1 Philippe Pasero 1 and Jérôme Moreaux 1,3,8 1IGH, CNRS, Université de Montpellier, Montpellier, France; 2Department of Hematology and Immunology, Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium; 3CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biologi - cal Hematology, Montpellier, France; 4CHU Montpellier, Department of Clinical Hematology, Montpellier, France; 5Medizinische Klinik und Poliklinik V, Universitätsklinikum Heidelberg, Heidelberg, Germany; 6Nationales Centrum für Tumorerkrankungen, Heidelberg , Ger - many; 7Université de Montpellier, UMR CNRS 5235, Montpellier, France and 8 Université de Montpellier, UFR de Médecine, Montpel - lier, France ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2018.208306 Received: October 5, 2018. Accepted: July 5, 2019. Pre-published: July 9, 2019. Correspondence: JEROME MOREAUX - [email protected] Supplementary experiment procedures Kinome Index A list of 661 genes of kinases or kinases related have been extracted from literature9, and challenged in the HM cohort for OS prognostic values The prognostic value of each of the genes was computed using maximally selected rank test from R package MaxStat. After Benjamini Hochberg multiple testing correction a list of 104 significant prognostic genes has been extracted. This second list has then been challenged for similar prognosis value in the UAMS-TT2 validation cohort.