Evolution and Structure of Clinically Relevant Gene Fusions in Multiple Myeloma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

The Drug Sensitivity and Resistance Testing (DSRT) Approach
A phenotypic screening and machine learning platform eciently identifies triple negative breast cancer-selective and readily druggable targets Prson Gautam 1 Alok Jaiswal 1 Tero Aittokallio 1, 2 Hassan Al Ali 3 Krister Wennerberg 1,4 Identifying eective oncogenic targets is challenged by the complexity of genetic alterations in 1Institute for Molecular Medicine Finland (FIMM), HiLIFE, University of Helsinki, Finland cancer and their poorly understood relation to cell function and survival. There is a need for meth- Current kinome coverage of kinase inhibitors in TNBC exhibit diverse kinase dependencies MFM-223 is selectively addicted to FGFR2 2Department of Mathematics and Statistics, University of Turku, Finland 3The Miami Project to Cure Paralysis, Peggy and Harold Katz Family Drug Discovery Center, A A Sylvester Comprehensive Cancer Center, and Department of Neurological Surgery and Medicine ods that rapidly and accurately identify “pharmacologically eective” targets without the require- clinical evaluation TN Kinases MFM-223 CAL-120 MDA-MB-231 TNBC TNBC TNBC TNBC TNBC TNBC HER2+ 100 University of Miami Miller School of Medicine, Miami, FL 33136, USA. non- HER2+ FGFR1 0.97 0.00 0.00 MFM-223 BL1 BL2 M MSL IM LAR ER+, PR+ 50 ment for priori knowledge of complex signaling networks. We developed an approach that uses ma- cancerous FGFR2 56.46 0.00 0.00 CAL-120 25 4 MDA-MB-231 Biotech Research & Innovation Centre (BRIC) and Novo Nordisk Foundation Center HCC1937 CAL-85-1 CAL-120 MDA-MB-231 DU4475 CAL-148 MCF-10A SK-BR-3 BT-474 FGFR3 25.10 0.00 0.00 0 chine learning to relate results from unbiased phenotypic screening of kinase inhibitors to their bio- for Stem Cell Biology (DanStem), University of Copenhagen, Denmark HCC1599 HDQ-P1 BT-549 MDA-MB-436 MFM-223 FGFR4 0.00 0.00 0.00 MAXIS*Bk Clinical status MDA-MB-468 CAL-51 Hs578T MDA-MB-453 score chemical activity data. -

Supplementary Information Material and Methods
MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Supplementary Information Material and methods Chemicals The EGFR inhibitor NVP-AEE788 (Novartis), the Jak inhibitor I (Merck Calbiochem, #420099) and anisomycin (Alomone labs, # A-520) were prepared as 50 mM stock solutions in 100% DMSO. Doxorubicin (Adriablastin, Pfizer), EGF (Sigma Ref: E9644), PDGF (Sigma, Ref: P4306) and IL-4 (Sigma, Ref: I-4269) stock solutions were prepared as recommended by the manufacturer. For in vivo administration: Temodal (20 mg Temozolomide capsules, Essex Chemie AG, Luzern) was dissolved in 4 mL KZI/glucose (20/80, vol/vol); Taxotere was bought as 40 mg/mL solution (Sanofi Aventis, France), and prepared in KZI/glucose. Antibodies The primary antibodies used were as follows: anti-S473P-Akt (#9271), anti-T308P-Akt (#9276,), anti-S9P-GSK3β (#9336), anti-T389P-p70S6K (#9205), anti-YP/TP-Erk1/2 (#9101), anti-YP/TP-p38 (#9215), anti-YP/TP-JNK1/2 (#9101), anti-Y751P-PDGFR (#3161), anti- p21Cip1/Waf1 (#2946), anti-p27Kip1 (#2552) and anti-Ser15-p53 (#9284) antibodies were from Cell Signaling Technologies; anti-Akt (#05-591), anti-T32P-FKHRL1 (#06-952) and anti- PDGFR (#06-495) antibodies were from Upstate; anti-IGF-1R (#SC-713) and anti-EGFR (#SC-03) antibodies were from Santa Cruz; anti-GSK3α/β (#44610), anti-Y641P-Stat6 (#611566), anti-S1981P-ATM (#200-301), anti-T2609 DNA-PKcs (#GTX24194) and anti- 1 MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Y1316P-IGF-1R were from Bio-Source International, Becton-Dickinson, Rockland, GenTex and internal production, respectively. The 4G10 antibody was from Millipore (#05-321MG). -
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

PRKCQ Promotes Oncogenic Growth and Anoikis Resistance of a Subset
Byerly et al. Breast Cancer Research (2016) 18:95 DOI 10.1186/s13058-016-0749-6 RESEARCH ARTICLE Open Access PRKCQ promotes oncogenic growth and anoikis resistance of a subset of triple- negative breast cancer cells Jessica Byerly1†, Gwyneth Halstead-Nussloch1†, Koichi Ito1, Igor Katsyv3 and Hanna Y. Irie1,2* Abstract Background: The protein kinase C (PKC) family comprises distinct classes of proteins, many of which are implicated in diverse cellular functions. Protein tyrosine kinase C theta isoform (PRKCQ)/PKCθ, a member of the novel PKC family, may have a distinct isoform-specific role in breast cancer. PKCθ is preferentially expressed in triple-negative breast cancer (TNBC) compared to other breast tumor subtypes. We hypothesized that PRKCQ/PKCθ critically regulates growth and survival of a subset of TNBC cells. Methods: To elucidate the role of PRKCQ/PKCθ in regulating growth and anoikis resistance, we used both gain and loss of function to modulate expression of PRKCQ. We enhanced expression of PKCθ (kinase-active or inactive) in non-transformed breast epithelial cells (MCF-10A) and assessed effects on epidermal growth factor (EGF)- independent growth, anoikis, and migration. We downregulated expression of PKCθ in TNBC cells, and determined effects on in vitro and in vivo growth and survival. TNBC cells were also treated with a small molecule inhibitor to assess requirement for PKCθ kinase activity in the growth of TNBC cells. Results: PRKCQ/PKCθ can promote oncogenic phenotypes when expressed in non-transformed MCF-10A mammary epithelial cells; PRKCQ/PKCθ enhances anchorage-independent survival, growth-factor-independent proliferation, and migration. PKCθ expression promotes retinoblastoma (Rb) phosphorylation and cell-cycle progression under growth factor-deprived conditions that typically induce cell-cycle arrest of MCF-10A breast epithelial cells. -

The Number of Genes
Table S1. The numbers of KD genes in each KD time The number The number The number The number Cell lines of genes of genes of genes of genes (96h) (120h) (144h) PC3 3980 3822 128 1725 A549 3724 3724 0 0 MCF7 3688 3471 0 1837 HT29 3665 3665 0 0 A375 3826 3826 0 0 HA1E 3801 3801 0 0 VCAP 4134 34 4121 0 HCC515 3522 3522 0 0 Table S2. The predicted results in the PC3 cell line on the LINCS II data id target rank A07563059 ADRB2 48 A12896037 ADRA2C 91 A13021932 YES1 77 PPM1B;PPP1CC;PPP2CA; A13254067 584;1326;297;171;3335 PTPN1;PPP2R5A A16347691 GMNN 2219 PIK3CB;MTOR;PIK3CA;PIK A28467416 18;10;9;13;8 3CG;PIK3CD A28545468 EHMT2;MAOB 14;67 A29520968 HSPB1 1770 A48881734 EZH2 1596 A52922642 CACNA1C 201 A64553394 ADRB2 155 A65730376 DOT1L 3764 A82035391 JUN 378 A82156122 DPP4 771 HRH1;HTR2C;CHRM3;CH A82772293 2756;2354;2808;2367 RM1 A86248581 CDA 1785 A92800748 TEK 459 A93093700 LMNA 1399 K00152668 RARB 105 K01577834 ADORA2A 525 K01674964 HRH1;BLM 31;1314 K02314383 AR 132 K03194791 PDE4D 30 K03390685 MAP2K1 77 K06762493 GMNN;APEX1 1523;2360 K07106112 ERBB4;ERBB2;EGFR 497;60;23 K07310275 AKT1;MTOR;PIK3CA 13;12;1 K07753030 RGS4;BLM 3736;3080 K08109215 BRD2;BRD3;BRD4 1413;2786;3 K08248804 XIAP 88 K08586861 TBXA2R;MBNL1 297;3428 K08832567 GMNN;CA12 2544;50 LMNA;NFKB1;APEX1;EH K08976401 1322;341;3206;123 MT2 K09372874 IMPDH2 232 K09711437 PLA2G2A 59 K10859802 GPR119 214 K11267252 RET;ALK 395;760 K12609457 LMNA 907 K13094524 BRD4 7 K13662825 CDK4;CDK9;CDK5;CDK1 34;58;13;18 K14704277 LMNA;BLM 1697;1238 K14870255 AXL 1696 K15170068 MAN2B1 1756 K15179879 -
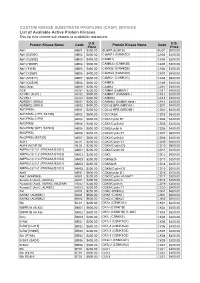
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This List May Change with Respect to Availability and Pricing
CUSTOM KINASE SUBSTRATE PROFILING (CKSP) SERVICE List of Available Active Protein Kinases This list may change with respect to availability and pricing. Protein Kinase Name Code U.S. Protein Kinase Name Code U.S. Price Price Abl1 AB01 $200.00 BUBR1(BUB1B) BU01 $600.00 Abl1 [E255K] AB02 $400.00 CaMK1δ (CAMK1D) CA03 $200.00 Abl1 [G250E] AB03 $400.00 CAMK1γ CA04 $200.00 Abl1 [H369P] AB04 $600.00 CAMK2α (CAMK2B) CA05 $200.00 Abl1 [T315I] AB05 $400.00 CaMK2β (CAMK2B) CA06 $200.00 Abl1 [Y253F] AB06 $400.00 CaMK2δ (CAMK2D) CA07 $400.00 Abl1 [M351T] AB07 $600.00 CaMK2γ (CAMK2G) CA08 $600.00 Abl1 [Q252H] AB08 $600.00 CAMK3γ CA09 $200.00 Abl2 (Arg) AB09 $200.00 CAMK4 CA10 $200.00 ACK AC01 $200.00 CAMK4 (CaMKIV ) CA11 $400.00 ACVR1 (ALK2 ) AC02 $400.00 CAMKK1 (CAMKKA ) CA12 $200.00 ACVRL1 AC03 $400.00 CAMKK2 CA13 $200.00 ADRBK1 (GRK2) AD01 $200.00 CAMKK2 (CaMKK beta ) CA14 $400.00 ADRBK2 (GRK3) AD02 $400.00 CDC42 BPA (MRCKA ) CD01 $400.00 Akt1/PKBα AK01 $200.00 CDC42 BPB (MRCKB ) CD02 $400.00 Akt1/PKBα [δPH, S473D] AK02 $600.00 CDC7/ASK CD03 $600.00 Akt1/PKBα [δPH] AK03 $600.00 CDK1/cyclin B1 CD04 $400.00 Akt2/PKBβ AK04 $200.00 CDK1/CyclinA2 CD05 $200.00 Akt2/PKBβ [δPH, S474D] AK05 $600.00 CDK2/cyclin A CD06 $400.00 Akt3/PKBγ AK06 $200.00 CDK2/Cyclin E1 CD07 $600.00 Akt3/PKBγ [S472D] AK07 $600.00 CDK2/CyclinA2 CD08 $200.00 ALK1 AL01 $200.00 CDK3/Cyclin E1 CD09 $600.00 ALK4 (ACVR1B) AL02 $200.00 CDK4//Cyclin D3 CD10 $600.00 AMPKα1/β1/γ1 (PRKAA1/B1/G1) AM01 $200.00 CDK4/Cyclin D1 CD11 $200.00 AMPKα1/β1/γ2 (PRKAA1/B1/G2) AM02 $200.00 CDK5 CD12 $600.00 -

Identification of DNA Response Elements Regulating Expression of CCAAT/Enhancer-Binding Protein (C/EBP) Β and Δ During Early A
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Identification of DNA response elements regulating expression of CCAAT/enhancer-binding protein (C/EBP) β and ẟ during early adipogenesis James E. Merrett1,2, Tao Bo1,4, Peter J. Psaltis1,3 and Christopher G. Proud1,2 * 1South Australian Health and Medical Research Institute, Lifelong Health, Adelaide, South Australia, Australia 2University of Adelaide, Department of Molecular and Biomedical Science, Adelaide, South Australia, Australia 3University of Adelaide, Adelaide Medical School, Adelaide, South Australia, Australia 4Shandong-South Australia Joint Laboratory of Metabolic Disease Research, Shandong Provincial Hospital affiliated to Shandong First Medical University *Corresponding author: Christopher Proud E-mail: [email protected] Running title: DNA response elements regulating early adipogenesis Keywords: adipogenesis, adipocyte, cyclic AMP (cAMP), cAMP response element-binding protein (CREB), CCAAT/enhancer-binding protein (C/EBP), glucocorticoid, glucocorticoid receptor, MAPK- interacting kinase (MNK) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.22.110114; this version posted May 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Given the high and increasing prevalence of obesity and associated disorders, such as type-2 diabetes, it is important to understand the mechanisms that regulate lipid storage and the differentiation of fat cells, a process termed adipogenesis. -

Lipid Metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives
cancers Review Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives 1, 1, 1 2 1 Laurence Pellerin y, Lorry Carrié y , Carine Dufau , Laurence Nieto , Bruno Ségui , 1,3 1, , 1, , Thierry Levade , Joëlle Riond * z and Nathalie Andrieu-Abadie * z 1 Centre de Recherches en Cancérologie de Toulouse, Equipe Labellisée Fondation ARC, Université Fédérale de Toulouse Midi-Pyrénées, Université Toulouse III Paul-Sabatier, Inserm 1037, 2 avenue Hubert Curien, tgrCS 53717, 31037 Toulouse CEDEX 1, France; [email protected] (L.P.); [email protected] (L.C.); [email protected] (C.D.); [email protected] (B.S.); [email protected] (T.L.) 2 Institut de Pharmacologie et de Biologie Structurale, CNRS, Université Toulouse III Paul-Sabatier, UMR 5089, 205 Route de Narbonne, 31400 Toulouse, France; [email protected] 3 Laboratoire de Biochimie Métabolique, CHU Toulouse, 31059 Toulouse, France * Correspondence: [email protected] (J.R.); [email protected] (N.A.-A.); Tel.: +33-582-7416-20 (J.R.) These authors contributed equally to this work. y These authors jointly supervised this work. z Received: 15 September 2020; Accepted: 23 October 2020; Published: 27 October 2020 Simple Summary: Melanoma is a devastating skin cancer characterized by an impressive metabolic plasticity. Melanoma cells are able to adapt to the tumor microenvironment by using a variety of fuels that contribute to tumor growth and progression. In this review, the authors summarize the contribution of the lipid metabolic network in melanoma plasticity and aggressiveness, with a particular attention to specific lipid classes such as glycerophospholipids, sphingolipids, sterols and eicosanoids.