(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
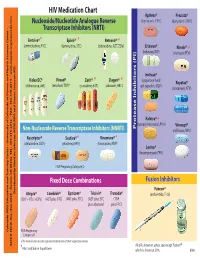
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 27, 2021, 11:42 pm Saquinavir (Invirase) Table of Contents Saquinavir Invirase Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Saquinavir, the first protease inhibitor approved by the FDA (in 1995), played a central part in the initial roll out of highly active combination antiretroviral therapy in the mid-1990’s. Three formulations of saquinavir have been approved, including the initial hard-gel capsule, a soft-gel capsule (now discontinued), and, more recently, a tablet. In the early years of combination antiretroviral therapy, unboosted saquinavir was used in combination with two nucleoside reverse transcriptase inhibitors (NRTIs). Subsequently, saquinavir has been used in combination with low-dose ritonavir boosting. Over time, multiple better-tolerated and more convenient antiretroviral agents have become available and have replaced saquinavir. At this time, saquinavir is no longer recommended for use in antiretroviral therapy regimens and patients taking this agent should switch to a currently recommended option. Key Clinical Trials Early trials demonstrated that saquinavir plus two NRTI’s (zidovudine and zalcitabine) led to greater reductions in HIV RNA and increases in CD4 count as compared to dual therapy with zidovudine plus saquinavir or zidovudine plus zalcitabine [ACTG 229]. Subsequently, saquinavir was compared to other protease inhibitors, each given with two NRTIs. One study demonstrated similar antiviral potency between saquinavir boosted with ritonavir and lopinavir-ritonavir, each given with tenofovir DF-emtricitabine [GEMINI], whereas a similar trial demonstrated higher rates of treatment failure and discontinuation in the saquinavir plus ritonavir arm and concluded that lopinavir-ritonavir has superior antiviral efficacy, largely driven by tolerability and patient preference [MaxCmin2]. -

SELZENTRY (Maraviroc) Tablets, for Oral Use • Hepatotoxicity Accompanied by Severe Rash Or Systemic Allergic Reaction
---------------------------------- CONTRAINDICATIONS --------------------------------- HIGHLIGHTS OF PRESCRIBING INFORMATION • SELZENTRY is contraindicated in patients with severe renal impairment These highlights do not include all the information needed to use or end-stage renal disease (ESRD) (CrCl less than 30 mL per minute) SELZENTRY safely and effectively. See full prescribing information for who are concomitantly taking potent CYP3A inhibitors or inducers. (4) SELZENTRY. -------------------------- WARNINGS AND PRECAUTIONS -------------------------- SELZENTRY (maraviroc) tablets, for oral use • Hepatotoxicity accompanied by severe rash or systemic allergic reaction, SELZENTRY (maraviroc) oral solution including potentially life-threatening events, has been reported. Hepatic Initial U.S. Approval: 2007 laboratory parameters including ALT, AST, and bilirubin should be obtained prior to starting SELZENTRY and at other time points during WARNING: HEPATOTOXICITY See full prescribing information for complete boxed warning. treatment as clinically indicated. If rash or symptoms or signs of hepatitis or allergic reaction develop, hepatic laboratory parameters should be • Hepatotoxicity has been reported which may be preceded by severe monitored and discontinuation of treatment should be considered. When rash or other features of a systemic allergic reaction (e.g., fever, administering SELZENTRY to patients with pre-existing liver eosinophilia, or elevated IgE). (5.1) dysfunction or who are co-infected with hepatitis B and/or C virus, • -

SYMTUZA™ (Darunavir, Cobicistat, Emtricitabine, Tenofovir Alafenamide) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 9/20/2018 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: SYMTUZA™ (darunavir, cobicistat, emtricitabine, tenofovir alafenamide) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. -

Treatment of AIDS and HIV-Related Conditions: 2001 J Am Board Fam Pract: First Published As on 1 July 2001
Current Report—HIV Treatment of AIDS and HIV-Related Conditions: 2001 J Am Board Fam Pract: first published as on 1 July 2001. Downloaded from Ronald H. Goldschmidt, MD, and Betty J. Dong, PharmD Managing human immunodeficiency virus (HIV) offer the best opportunity to provide comprehen- disease and the acquired immunodeficiency syn- sive care. drome (AIDS) has become more standardized yet This Current Report—HIV updates our annual more complex during the past year. Antiretroviral treatment guidelines.3 These recommendations treatment guidelines now represent a general con- (Table 1) are based on our experience at San Fran- sensus on basic treatment principles and options1 as cisco General Hospital, published guidelines, a re- benefits and risks of therapy have become more view of the medical literature, and experience evident. Clinical manifestations of HIV infection, gained from answering telephone calls to our Na- prognosis, and quality of life are clearly improved tional HIV Telephone Consultation Service for most patients receiving potent antiretroviral (Warmline). Because HIV management changes therapy, yet viral resistance and chronic and short- rapidly, clinicians are advised to refer to the excel- term drug toxicities remain major problems. The lent federal guidelines for the use of antiretroviral markedly decreased incidence of opportunistic in- agents in HIV-infected adults and adolescents1 and fections among treated patients has made unusual for prevention of opportunistic infections,4 which infections less common in daily HIV management. are updated frequently on the Internet (Table 2). The role of resistance assays in HIV care is still These and other federal guidelines are available at being assessed, but they offer great possibilities for http://www.hivatis.org. -

(Lupin Limited), HA659 WHOPAR Part 4 US Prescribing Information March 2016 Page 1 of 23
Lamivudine/Zidovudine 150mg/300mg WHOPAR part 4 March 2016 Tablets US prescribing information (Lupin Limited), HA659 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use ---------------------- WARNINGS AND PRECAUTIONS ---------------------- . lamivudine and zidovudine tablets safely and effectively. See full See boxed warning for information about the following: hematologic toxicity, symptomatic myopathy, lactic acidosis and severe prescribing information for lamivudine and zidovudine tablets. hepatomegaly, and severe acute exacerbations of hepatitis B. (5.1, 5.2, Lamivudine and Zidovudine Tablets USP, 150 mg/300 mg 5.3, 5.4) . Lamivudine and zidovudine tablets should not be administered with Initial U.S. Approval: 1997 other lamivudine- or zidovudine-containing products or emtricitabine- containing products. (5.5) WARNING: RISK OF HEMATOLOGIC TOXICITY, MYOPATHY, . Hepatic decompensation, some fatal, has occurred in HIV-1/HCV co- LACTIC ACIDOSIS, EXACERBATIONS OF HEPATITIS B infected patients receiving combination antiretroviral therapy and See full prescribing information for complete boxed warning. interferon alfa with/without ribavirin. Discontinue lamivudine and • Hematologic toxicity including neutropenia and anemia have been zidovudine tablets as medically appropriate and consider dose reduction associated with the use of zidovudine, one of the components of or discontinuation of interferon alfa, ribavirin, or both. (5.6) lamivudine and zidovudine tablets. (5.1) . Exacerbation of anemia has been reported in HIV-1/HCV co-infected • Symptomatic myopathy associated with prolonged use of zidovudine. patients receiving ribavirin and zidovudine. Co-administration of (5.2) ribavirin and zidovudine is not advised. (5.6) • Lactic acidosis and hepatomegaly with steatosis, including fatal cases, . Pancreatitis: Use with caution in pediatric patients with a history of have been reported with the use of nucleoside analogues including pancreatitis or other significant risk factors for pancreatitis. -

Association Among Ccr5 Genotypes, Ccr5 Expression, and In
ASSOCIATION AMONG CCR5 GENOTYPES, CCR5 EXPRESSION, AND IN VITRO HIV INFECTION Bangan John Submitted in partial fulfilment of the requirements For the degree of Master of Science Dissertation Advisor: Dr Peter A. Zimmerman Department of Biology CASE WESTERN RESERVE UNIVERSITY May, 2013 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Bangan John ______________________________________________________ Master of Science candidate for the ________________________________degree *. Roy Ritzmann (signed)_______________________________________________ (chair of the committee) Peter Zimmerman ________________________________________________ Christopher Cullis ________________________________________________ Daniel Tisch ________________________________________________ 12/20/2012 (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Dedication: I would like to dedicate this thesis to my family, the John’s. Table of Contents List of Tables ................................................................................................................. ii List of Figures .............................................................................................................. iii Acknowledgements ....................................................................................................... iv Abbreviations ................................................................................................................ -

Product Monograph for KIVEXA
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrKIVEXA abacavir and lamivudine tablets 600 mg abacavir (as abacavir sulfate) and 300 mg lamivudine, Oral Antiretroviral Agent ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Initial Approval: July 25, 2005 Date of Revision: September 21, 2021 Submission Control No: 243500 ©2021 ViiV Healthcare group of companies or its licensor Trademarks are owned by or licensed to the ViiV Healthcare group of companies Page 1 of 44 RECENT MAJOR LABEL CHANGES 4 Dosage and Administration, 4.1 Dosing Considerations 08/2020 3 Serious Warnings and Precautions Box 05/2021 7 Warnings and Precautions, General, Renal Insufficiency 08/2020 7 Warnings and Precautions, Clinical Management of Abacavir HSRs 05/2021 7 Warnings and Precautions, Immune 05/2021 TABLE OF CONTENTS RECENT MAJOR LABEL CHANGES......................................................................................2 TABLE OF CONTENTS ..............................................................................................................2 PART I: HEALTH PROFESSIONAL INFORMATION............................................................4 1 INDICATIONS....................................................................................................................4 1.1 Pediatrics (< 18 years of age).....................................................................................4 1.2 Geriatrics (≥ 65 years of age).....................................................................................4 -

REYATAZ Rx Only
® REYATAZ Rx only (atazanavir sulfate) Capsules (Patient Information Leaflet Included) DESCRIPTION REYATAZ® (atazanavir sulfate) is an azapeptide inhibitor of HIV-1 protease. The chemical name for atazanavir sulfate is (3S,8S,9S,12S)-3,12-Bis(1,1- dimethylethyl)-8-hydroxy-4,11-dioxo-9-(phenylmethyl)-6-[[4-(2-pyridinyl)phenyl]methyl]- 2,5,6,10,13-pentaazatetradecanedioic acid dimethyl ester, sulfate (1:1). Its molecular formula is C38H52N6O7•H2SO4, which corresponds to a molecular weight of 802.9 (sulfuric acid salt). The free base molecular weight is 704.9. Atazanavir sulfate has the following structural formula: Atazanavir sulfate is a white to pale yellow crystalline powder. It is slightly soluble in water (4-5 mg/mL, free base equivalent) with the pH of a saturated solution in water being about 1.9 at 24 ± 3° C. REYATAZ Capsules are available for oral administration in strengths containing the equivalent of 100 mg, 150 mg, or 200 mg of atazanavir as atazanavir sulfate and the following inactive ingredients: crospovidone, lactose monohydrate, and magnesium stearate. The capsule shells contain the following inactive ingredients: gelatin, FD&C Blue #2, and titanium dioxide. The capsules are printed with ink containing shellac, titanium dioxide, FD&C Blue #2, isopropyl alcohol, ammonium hydroxide, propylene glycol, n-butyl alcohol, simethicone, and dehydrated alcohol. 1 of 47 Approved v1.0 CLINICAL PHARMACOLOGY Microbiology Mechanism of Action Atazanavir (ATV) is an azapeptide HIV-1 protease inhibitor (PI). The compound selectively inhibits the virus-specific processing of viral Gag and Gag-Pol polyproteins in HIV-1 infected cells, thus preventing formation of mature virions. -

Drug Class Review
Drug Class Review HIV Protease Inhibitors 8:18.08.08 HIV Protease Inhibitors Atazanavir (Reyataz®) Atazanavir/Cobicistat (Evotaz®) Darunavir (Prezista®) Darunavir/Cobicistat (Prexcobix®) Fosamprenavir (Lexiva®) Indinavir (Crixivan®) Lopinavir/Ritonavir (Kaletra®) Nelfinavir (Viracept®) Ritonavir (Norvir®) Saquinavir (Invirase®) Tipranavir (Aptivus®) Final Report August 2015 Review prepared by: Melissa Archer, PharmD, Clinical Pharmacist Natalia Ruiz, PharmD Candidate 2016 Candice Nielson, PharmD Candidate 2016 Devin Stock, PharmD Candidate 2016 Gary Oderda, PharmD, MPH, Professor University of Utah College of Pharmacy Copyright © 2015 by University of Utah College of Pharmacy Salt Lake City, Utah. All rights reserved. 1 Table of Contents Executive Summary ......................................................................................................................... 3 Introduction .................................................................................................................................... 5 Table 1. Comparison of the HIV Protease Inhibitor Agents ................................................... 6 Disease Overview ...................................................................................................................... 11 Table 2. Summary of HIV Treatments .................................................................................. 12 Table 3. HIV/AIDS Disease Staging Systems ......................................................................... 21 Table 4. Most Current Clinical Practice -

Atazanavir Sulfate Formulations with Improved
(19) TZZ _T (11) EP 2 555 757 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 9/20 (2006.01) A61K 31/4402 (2006.01) 25.05.2016 Bulletin 2016/21 (86) International application number: (21) Application number: 11714907.0 PCT/US2011/031526 (22) Date of filing: 07.04.2011 (87) International publication number: WO 2011/127244 (13.10.2011 Gazette 2011/41) (54) ATAZANAVIR SULFATE FORMULATIONS WITH IMPROVED PH EFFECT ATAZANAVIR-SULFAT-FORMULIERUNGEN MIT VERBESSERTEM PH-EFFEKT FORMULATIONS DE SULFATE D’ATAZANAVIR AVEC UN EFFET PH AMÉLIORÉ (84) Designated Contracting States: • QIAN, Feng AL AT BE BG CH CY CZ DE DK EE ES FI FR GB New Brunswick GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO New Jersey 08903 (US) PL PT RO RS SE SI SK SM TR (74) Representative: Reitstötter Kinzebach (30) Priority: 09.04.2010 US 322487 P Patentanwälte Sternwartstrasse 4 (43) Date of publication of application: 81679 München (DE) 13.02.2013 Bulletin 2013/07 (56) References cited: (73) Proprietor: Bristol-Myers Squibb Holdings Ireland WO-A1-95/09614 WO-A1-2005/034948 6312 Steinhausen (CH) WO-A2-2009/084036 (72) Inventors: • Karen Dahri ET AL: "Atazanavir and • NIKFAR, Faranak Acid-Lowering Therapy", Canadian Journal of New Brunswick Hospital Pharmacy, 1 January 2008 (2008-01-01), New Jersey 08903 (US) pages 21-29, XP55021068, Retrieved from the • HUSSAIN, Munir Alwan Internet: New Brunswick URL:http://www.cjhp-online.ca/index.php/cj New Jersey 08903 (US) hp/article/viewFile/7/6# [retrieved on 2012-03-06] Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Antiviral Drugs in the Treatment of Aids: What Is in the Pipeline ?
October 15, 2007 EU RO PE AN JOUR NAL OF MED I CAL RE SEARCH 483 Eur J Med Res (2007) 12: 483-495 © I. Holzapfel Publishers 2007 ANTIVIRAL DRUGS IN THE TREATMENT OF AIDS: WHAT IS IN THE PIPELINE ? Hans-Jürgen Stellbrink Infektionsmedizinisches Centrum Hamburg ICH, Hamburg, Germany Abstract even targeting immune responses have to be devel- Drug development in the field of HIV treatment is oped. Continued drug development serves the ulti- rapid. New nucleoside analogues (NRTI), non-nucleo- mate goal of normalization of life expectancy. side analogue reverse transcriptase inhibitors (NNR- TI), and protease inhibitors (PI) are currently being in- METHODS vestigated in human trials. Furthermore, inhibitors of HIV attachment, fusion and integrase with novel Drug development in the field of HIV infection is modes of action are being developed, which offer new highly competitive, and a company’s decision to pur- perspectives for the goal of a normalization of life-ex- sue or discontinue the development of a drug is dri- pectancy in HIV-infected individuals. The most ad- ven by economic rather than scientific considerations. vanced compounds likely to become licensed soon in- Of all candidate compounds, only a few reach the lev- clude the NNRTIs rilpivirine and etravirine, the inte- el of trials in humans, and some exhibit lack of effica- grase inhibitors raltegravir and elvitegravir, and mar- cy or toxicity problems at this stage. Some compounds aviroc and vicriviroc, novel inhibitors of the CCR5 also have no obvious advantage over currently avail- chemokine receptor, which functions as the major able ones, so that their development is discontinued.