Total Synthesis of Thia-Calanolide a and Analogues, Asymmetric Aziridination of Alkenes and Development of Environmentally Benign Green Methodologies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Treatment-Drugs
© National HIV Curriculum PDF created September 23, 2021, 9:14 am Stavudine (Zerit) Table of Contents Stavudine Zerit Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Stavudine, an early nucleoside reverse transcriptase inhibitor (NRTI), was used as part of combination antiretroviral therapy for years, but now has become obsolete and replaced by better-tolerated and safer options. Stavudine poses risk of serious toxicity, including peripheral neuropathy (which can be permanent), pancreatitis, lipoatrophy, and lactic acidosis. Fatal and nonfatal cases of pancreatitis and lactic acidosis have been reported, especially when stavudine was combined with didanosine. According to the Adult and Adolescent ARV Guidelines, stavudine is no longer recommended for the treatment of HIV infection due to potential severe toxicity. Further, all persons currently taking stavudine should be strongly encouraged to switch to a safer medication. The sale and distribution of all strengths of stavudine will be discontinued and removed from the market in the United States in 2020. Key Clinical Trials Stavudine was studied for treatment-naïve patients as part of triple therapy, such as with lamivudine plus indinavir [START I], lamivudine plus lopinavir-ritonavir [M98-863], and lamivudine plus efavirenz [DART II]. Several studies demonstrated benefits of switching stavudine to newer NRTI agents, such as tenofovir disoproxil fumarate; the switch led to decreased rates of metabolic complications and mitochondrial toxicity [903E, SNAP, and ACTG 5142]. Resistance For a listing of the most common clinically significant mutations associated with stavudine (d4T) resistance, see the NRTI Resistance Notes on the Stanford University HIV Drug Resistance Database. Page 1/2 Key Drug Interactions For complete information on stavudine-related drug interactions, see the Drug Interactions section in the Stavudine (Zerit) Prescribing Information. -

Eparate Formulations According to the Prescribed Dosing Recommendations for These Products
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Lamivudine/Zidovudine Teva 150 mg/300 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each film-coated tablet contains 150 mg lamivudine and 300 mg zidovudine. For the full list of excipients see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet White, capsule shaped, biconvex, film-coated scored tablet – engraved with “L/Z” on one side and “150/300” on the other side. The tablet can be divided into equal halves. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Lamivudine/Zidovudine Teva is indicated in antiretroviral combination therapy for the treatment of Human Immunodeficiency Virus (HIV) infection (see section 4.2). 4.2 Posology and method of administration Therapy should be initiated by a physician experienced in the management of HIV infection. Lamivudine/Zidovudine Teva may be administered with or without food. To ensure administration of the entire dose, the tablet(s) should ideally be swallowed without crushing. For patients who are unable to swallow tablets, tablets may be crushed and added to a small amount of semi-solid food or liquid, all of which should be consumed immediately (see section 5.2). Adults and adolescents weighing at least 30 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one tablet twice daily. Children weighing between 21 kg and 30 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one-half tablet taken in the morning and one whole tablet taken in the evening. Children weighing from 14 kg to 21 kg: the recommended oral dose of Lamivudine/Zidovudine Teva is one-half tablet taken twice daily. -
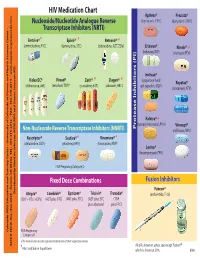
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

TRIZIVIR® (Abacavir Sulfate, Lamivudine, and Zidovudine) Tablets
NDA 21-205/S-011 Page 4 PRESCRIBING INFORMATION TRIZIVIR® (abacavir sulfate, lamivudine, and zidovudine) Tablets WARNINGS TRIZIVIR contains 3 nucleoside analogues (abacavir sulfate, lamivudine, and zidovudine) and is intended only for patients whose regimen would otherwise include these 3 components. Hypersensitivity Reactions: Serious and sometimes fatal hypersensitivity reactions have been associated with abacavir sulfate, a component of TRIZIVIR. Hypersensitivity to abacavir is a multi-organ clinical syndrome usually characterized by a sign or symptom in 2 or more of the following groups: (1) fever, (2) rash, (3) gastrointestinal (including nausea, vomiting, diarrhea, or abdominal pain), (4) constitutional (including generalized malaise, fatigue, or achiness), and (5) respiratory (including dyspnea, cough, or pharyngitis). Discontinue TRIZIVIR as soon as a hypersensitivity reaction is suspected. Permanently discontinue TRIZIVIR if hypersensitivity cannot be ruled out, even when other diagnoses are possible. Following a hypersensitivity reaction to abacavir, NEVER restart TRIZIVIR or any other abacavir-containing product because more severe symptoms can occur within hours and may include life-threatening hypotension and death. Reintroduction of TRIZIVIR or any other abacavir-containing product, even in patients who have no identified history or unrecognized symptoms of hypersensitivity to abacavir therapy, can result in serious or fatal hypersensitivity reactions. Such reactions can occur within hours (see WARNINGS and PRECAUTIONS: Information -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

ZIAGEN Safely and Effectively
HIGHLIGHTS OF PRESCRIBING INFORMATION • Patients With Hepatic Impairment: Mild hepatic impairment – 200 mg These highlights do not include all the information needed to use twice daily; moderate/severe hepatic impairment – contraindicated. (2.3) ZIAGEN safely and effectively. See full prescribing information for --------------------- DOSAGE FORMS AND STRENGTHS -------------- ZIAGEN. Tablets: 300 mg; Oral Solution: 20 mg/mL (3) ZIAGEN® (abacavir sulfate) Tablets and Oral Solution -------------------------------CONTRAINDICATIONS------------------------ Initial U.S. Approval: 1998 • Previously demonstrated hypersensitivity to abacavir. (4, 5.1) • Moderate or severe hepatic impairment. (4) WARNING: HYPERSENSITIVITY REACTIONS/LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY ----------------------- WARNINGS AND PRECAUTIONS ---------------- • Hypersensitivity: Serious and sometime fatal hypersensitivity reactions See full prescribing information for complete boxed warning. have been associated with ZIAGEN and other abacavir-containing • Serious and sometimes fatal hypersensitivity reactions have been products. Read full prescribing information section 5.1 before associated with ZIAGEN (abacavir sulfate). (5.1) prescribing ZIAGEN. (5.1) • Hypersensitivity to abacavir is a multi-organ clinical syndrome. • Lactic acidosis and severe hepatomegaly with steatosis have been (5.1) reported with the use of nucleoside analogues. (5.2) • Patients who carry the HLA-B*5701 allele are at high risk for • Immune reconstitution syndrome (5.3) and redistribution/accumulation -

Zero Dollar Cost Share – Generic Tenofovir Disoproxil Fumarate 300 Mg P&T Approval Date 11/2019, 8/2020, 9/2020 Effective Date 10/1/2020; Oxford Only: 11/1/2020
UnitedHealthcare Pharmacy Clinical Pharmacy Programs Program Number 2020 P 1289-3 Program Prior Authorization/Regulatory Medication HIV Pre-exposure Prophylaxis (PrEP) Zero Dollar Cost Share – generic tenofovir disoproxil fumarate 300 mg P&T Approval Date 11/2019, 8/2020, 9/2020 Effective Date 10/1/2020; Oxford only: 11/1/2020 . 1. Background: The U.S. Preventive Services Task Force (USPSTF) recommends that clinicians offer preexposure prophylaxis (PrEP) with effective antiretroviral therapy to persons who are at high risk of HIV acquisition.1 Once-daily oral treatment with Truvada® (emtricitabine/tenofovir disoproxil fumarate) is approved by the US Food and Drug Administration (FDA) for use as PrEP in persons at risk of sexual acquisition of HIV infection. Several studies reviewed by the USPSTF found that tenofovir disoproxil fumarate alone was also effective as PrEP and CDC guidelines note that, given these trial data, tenofovir disoproxil fumarate alone can be considered as an alternative regimen for high-risk heterosexually active men and women and persons who inject drugs.1-3 This program is designed to meet Health Care Reform requirements which require coverage of effective antiretroviral therapy, which includes tenofovir disoproxil fumarate or Truvada, at zero dollar cost share if being used for PrEP and criteria are met. 2. Coverage Criteria: A. Coverage at zero dollar cost share of generic tenofovir disoproxil fumarate 300 mg will be approved based on both of the following criteria: 1. Member is taking generic tenofovir disoproxil fumarate 300 mg as effective antiretroviral therapy for PrEP -AND- 2. Generic tenofovir disoproxil fumarate 300 mg will be used as part of a comprehensive prevention strategy including other prevention measures Authorization will be issued for zero copay with deductible bypass for 12 months. -

Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis
Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis Centers for Disease Control and Prevention Office of Infectious Diseases National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention Division of Tuberculosis Elimination June 2013 This document is accessible online at http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Suggested citation: CDC. Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis [online]. 2013. Available from URL: http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm Table of Contents Introduction 1 Methodology for Preparation of these Guidelines 2 The Role of Rifamycins in Tuberculosis Treatment 4 Managing Drug Interactions with Antivirals and Rifampin 5 Managing Drug Interactions with Antivirals and Rifabutin 9 Treatment of Latent TB Infection with Rifampin or Rifapentine 10 Treating Pregnant Women with Tuberculosis and HIV Co-infection 10 Treating Children with HIV-associated Tuberculosis 12 Co-treatment of Multidrug-resistant Tuberculosis and HIV 14 Limitations of these Guidelines 14 HIV-TB Drug Interaction Guideline Development Group 15 References 17 Table 1a. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in adults 21 Table 1b. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in children 22 Table 2a. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in adults 23 Table 2b. Recommendations for co-administering antiretroviral drugs with RIFAMPIN in children 25 Table 3. Recommendations for co-administering antiretroviral drugs with RIFABUTIN in adults 26 ii Introduction Worldwide, tuberculosis is the most common serious opportunistic infection among people with HIV infection. -

DESCOVY, and Upon Diagnosis of These Highlights Do Not Include All the Information Needed to Use Any Other Sexually Transmitted Infections (Stis)
HIGHLIGHTS OF PRESCRIBING INFORMATION once every 3 months while taking DESCOVY, and upon diagnosis of These highlights do not include all the information needed to use any other sexually transmitted infections (STIs). (2.2) DESCOVY safely and effectively. See full prescribing information • Recommended dosage: for DESCOVY. • Treatment of HIV-1 Infection: One tablet taken once daily with or ® without food in patients with body weight at least 25 kg. (2.3) DESCOVY (emtricitabine and tenofovir alafenamide) tablets, for • HIV-1 PrEP: One tablet taken once daily with or without food in oral use individuals with body weight at least 35 kg. (2.4) Initial U.S. Approval: 2015 • Renal impairment: DESCOVY is not recommended in individuals with WARNING: POST-TREATMENT ACUTE EXACERBATION OF estimated creatinine clearance below 30 mL per minute. (2.5) HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE ----------------------DOSAGE FORMS AND STRENGTHS-------------------- OF DESCOVY FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS Tablets: 200 mg of FTC and 25 mg of TAF (3) (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION See full prescribing information for complete boxed warning. -------------------------------CONTRAINDICATIONS------------------------------ DESCOVY for HIV-1 PrEP is contraindicated in individuals with Severe acute exacerbations of hepatitis B (HBV) have been unknown or positive HIV-1 status. (4) reported in HBV-infected individuals who have discontinued products containing emtricitabine (FTC) and/or tenofovir -----------------------WARNINGS AND PRECAUTIONS----------------------- disoproxil fumarate (TDF), and may occur with • Comprehensive management to reduce the risk of sexually discontinuation of DESCOVY. Hepatic function should be transmitted infections (STIs), including HIV-1, when DESCOVY is monitored closely in these individuals. -

Lamivudine in Combination with Zidovudine, Stavudine, Or Didanosine in Patients with HIV-1 Infection
Lamivudine in combination with zidovudine, stavudine, or didanosine in patients with HIV-1 infection. A randomized, double-blind, placebo-controlled trial Daniel R. Kuritzkes*, Ian Marschner†, Victoria A. Johnson‡, Roland Bassett†, Joseph J. Eron§, Margaret A. FischlII, Robert L. Murphy¶, Kenneth Fife**, Janine Maenza††, Mary E. Rosandich*, Dawn Bell‡‡, Ken Wood§§, Jean-Pierre Sommadossi‡, Carla PettinelliII II and the National Institute of Allergy and Infectious Disease AIDS Clinical Trials Group Protocol 306 Investigators Objective: To study the antiviral activity of lamivudine (3TC) plus zidovudine (ZDV), didanosine (ddI), or stavudine (d4T). Design: Randomized, placebo-controlled, partially double-blinded multicenter study. Setting: Adult AIDS Clinical Trials Units. Patients: Treatment-naive HIV-infected adults with 200–600 × 106 CD4 T lymphocytes/l. Interventions: Patients were openly randomized to a d4T or a ddI limb, then randomized in a blinded manner to receive: d4T (80 mg/day), d4T plus 3TC (300 mg/day), or ZDV (600 mg/day) plus 3TC, with matching placebos; or ddI (400 mg/day), ddI plus 3TC (300 mg/day), or ZDV (600 mg/day) plus 3TC, with matching placebos. After 24 weeks 3TC was added for patients assigned to the monotherapy arms. Main outcome measure: The reduction in plasma HIV-1 RNA level at weeks 24 and 48. From the *University of Colorado Health Sciences Center and Veterans Affairs Medical Center, Denver, Colorado, the †Center for Biostatistics in AIDS Research, Harvard School of Public Health, Boston, Massachusetts, the