HIVID® (Zalcitabine) Tablets - PACKAGE INSERT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
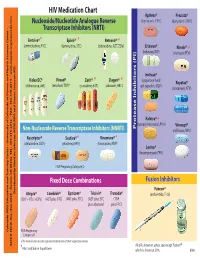
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

DESCOVY, and Upon Diagnosis of These Highlights Do Not Include All the Information Needed to Use Any Other Sexually Transmitted Infections (Stis)
HIGHLIGHTS OF PRESCRIBING INFORMATION once every 3 months while taking DESCOVY, and upon diagnosis of These highlights do not include all the information needed to use any other sexually transmitted infections (STIs). (2.2) DESCOVY safely and effectively. See full prescribing information • Recommended dosage: for DESCOVY. • Treatment of HIV-1 Infection: One tablet taken once daily with or ® without food in patients with body weight at least 25 kg. (2.3) DESCOVY (emtricitabine and tenofovir alafenamide) tablets, for • HIV-1 PrEP: One tablet taken once daily with or without food in oral use individuals with body weight at least 35 kg. (2.4) Initial U.S. Approval: 2015 • Renal impairment: DESCOVY is not recommended in individuals with WARNING: POST-TREATMENT ACUTE EXACERBATION OF estimated creatinine clearance below 30 mL per minute. (2.5) HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE ----------------------DOSAGE FORMS AND STRENGTHS-------------------- OF DESCOVY FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS Tablets: 200 mg of FTC and 25 mg of TAF (3) (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION See full prescribing information for complete boxed warning. -------------------------------CONTRAINDICATIONS------------------------------ DESCOVY for HIV-1 PrEP is contraindicated in individuals with Severe acute exacerbations of hepatitis B (HBV) have been unknown or positive HIV-1 status. (4) reported in HBV-infected individuals who have discontinued products containing emtricitabine (FTC) and/or tenofovir -----------------------WARNINGS AND PRECAUTIONS----------------------- disoproxil fumarate (TDF), and may occur with • Comprehensive management to reduce the risk of sexually discontinuation of DESCOVY. Hepatic function should be transmitted infections (STIs), including HIV-1, when DESCOVY is monitored closely in these individuals. -

Lamivudine in Combination with Zidovudine, Stavudine, Or Didanosine in Patients with HIV-1 Infection
Lamivudine in combination with zidovudine, stavudine, or didanosine in patients with HIV-1 infection. A randomized, double-blind, placebo-controlled trial Daniel R. Kuritzkes*, Ian Marschner†, Victoria A. Johnson‡, Roland Bassett†, Joseph J. Eron§, Margaret A. FischlII, Robert L. Murphy¶, Kenneth Fife**, Janine Maenza††, Mary E. Rosandich*, Dawn Bell‡‡, Ken Wood§§, Jean-Pierre Sommadossi‡, Carla PettinelliII II and the National Institute of Allergy and Infectious Disease AIDS Clinical Trials Group Protocol 306 Investigators Objective: To study the antiviral activity of lamivudine (3TC) plus zidovudine (ZDV), didanosine (ddI), or stavudine (d4T). Design: Randomized, placebo-controlled, partially double-blinded multicenter study. Setting: Adult AIDS Clinical Trials Units. Patients: Treatment-naive HIV-infected adults with 200–600 × 106 CD4 T lymphocytes/l. Interventions: Patients were openly randomized to a d4T or a ddI limb, then randomized in a blinded manner to receive: d4T (80 mg/day), d4T plus 3TC (300 mg/day), or ZDV (600 mg/day) plus 3TC, with matching placebos; or ddI (400 mg/day), ddI plus 3TC (300 mg/day), or ZDV (600 mg/day) plus 3TC, with matching placebos. After 24 weeks 3TC was added for patients assigned to the monotherapy arms. Main outcome measure: The reduction in plasma HIV-1 RNA level at weeks 24 and 48. From the *University of Colorado Health Sciences Center and Veterans Affairs Medical Center, Denver, Colorado, the †Center for Biostatistics in AIDS Research, Harvard School of Public Health, Boston, Massachusetts, the -

(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
US 20080241135A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson et al. (43) Pub. Date: Oct. 2, 2008 (54) METHODS FOR REDUCINGVIRAL LOAD IN 60/711,528, filed on Aug. 26, 2005, provisional appli HIV-1-INFECTED PATIENTS cation No. 60/715,619, filed on Sep. 9, 2005. (75) Inventors: William C. Olson, Ossining, NY Publication Classification (US); Paul J. Maddon, Scarsdale, NY (US); Daniel C. Pevear, (51) Int. Cl. Medford, MA (US); Robert J. A 6LX 39/395 (2006.01) Israel, Suffern, NY (US); Jose D. A6IP 43/00 (2006.01) Murga, Rosedale, NY (US) (52) U.S. Cl. ..................................................... 424/133.1 Correspondence Address: (57) ABSTRACT COOPER & DUNHAM, LLP 1185 AVENUE OF THE AMERICAS This method provides a method for reducing HIV-1 viral load NEW YORK, NY 10036 in an HIV-1-infected human subject which comprises admin istering to the subject at a predefined interval effective HIV-1 (73) Assignee: Progenics Pharmaceuticals, Inc. viral load-reducing doses of (a) a humanized antibody desig nated PRO 140, or of (b) an anti-CCR5 receptor monoclonal (21) Appl. No.: 11/894,762 antibody. This invention also provides a method for inhibiting in a human Subject the onset or progression of an HIV-1- (22) Filed: Aug. 20, 2007 associated disorder, the inhibition of which is effected by inhibiting fusion of HIV-1 to CCR5"CD4" target cells in the Related U.S. Application Data Subject. This invention also provides a method for treating a subject infected with HIV-1 comprising administering to the (63) Continuation of application No. -

DESCOVY® Formulary Monograph
DESCOVY® Formulary Monograph Click here for full Prescribing Information for DESCOVY, including BOXED WARNING. TABLE OF CONTENTS EXECUTIVE SUMMARY BOXED WARNING . 3 Indication . 3 Dosage and administration . 3 Adverse reactions . 4 Manufacturer . 4 Dosage forms and how supplied . 4 Wholesale acquisition cost (WAC) . 4 FDA approval date . 4 Clinical data summary . 5 HIV DISEASE STATE OVERVIEW The HIV patient profile . 7 The importance of early treatment . 7 Treatment overview . 8 DESCOVY SUMMARY OF PRODUCT INFORMATION BOXED WARNING . 9 Indication and usage . 9 Dosage and administration . 9 Product description and dosage forms . 10 Warnings and precautions . 10 Lactic acidosis/severe hepatomegaly with steatosis . 10 Severe acute exacerbation of hepatitis B in patients coinfected with HIV-1 and HBV . 10 Fat redistribution . 11 Immune reconstitution syndrome . 11 New onset or worsening renal impairment . 11 Bone loss and mineralization defects . 12 Adverse reactions . 12 Adverse reactions in clinical trials of FTC+TAF with EVG+COBI in treatment-naïve adults with HIV-1 infection . 12 Renal laboratory tests . 13 Bone mineral density effects . 13 Adverse reactions in clinical trials in pediatric patients with HIV-1 infection . 13 Drug interactions . 14 Potential for other drugs to affect one or more components of DESCOVY . 14 Drugs affecting renal function . 14 Established and other potentially significant drug interactions . 14 Drugs without clinically significant interactions with DESCOVY . 15 Click here for full Prescribing Information for DESCOVY, including BOXED WARNING. 1 TABLE OF CONTENTS DESCOVY SUMMARY OF PRODUCT INFORMATION (cont .) Use in specific populations . 15 Pregnancy . 15 Pregnancy exposure registry . 15 Risk summary . 15 Human data . 15 Lactation . 16 Risk summary . -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 27, 2021, 11:42 pm Saquinavir (Invirase) Table of Contents Saquinavir Invirase Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Saquinavir, the first protease inhibitor approved by the FDA (in 1995), played a central part in the initial roll out of highly active combination antiretroviral therapy in the mid-1990’s. Three formulations of saquinavir have been approved, including the initial hard-gel capsule, a soft-gel capsule (now discontinued), and, more recently, a tablet. In the early years of combination antiretroviral therapy, unboosted saquinavir was used in combination with two nucleoside reverse transcriptase inhibitors (NRTIs). Subsequently, saquinavir has been used in combination with low-dose ritonavir boosting. Over time, multiple better-tolerated and more convenient antiretroviral agents have become available and have replaced saquinavir. At this time, saquinavir is no longer recommended for use in antiretroviral therapy regimens and patients taking this agent should switch to a currently recommended option. Key Clinical Trials Early trials demonstrated that saquinavir plus two NRTI’s (zidovudine and zalcitabine) led to greater reductions in HIV RNA and increases in CD4 count as compared to dual therapy with zidovudine plus saquinavir or zidovudine plus zalcitabine [ACTG 229]. Subsequently, saquinavir was compared to other protease inhibitors, each given with two NRTIs. One study demonstrated similar antiviral potency between saquinavir boosted with ritonavir and lopinavir-ritonavir, each given with tenofovir DF-emtricitabine [GEMINI], whereas a similar trial demonstrated higher rates of treatment failure and discontinuation in the saquinavir plus ritonavir arm and concluded that lopinavir-ritonavir has superior antiviral efficacy, largely driven by tolerability and patient preference [MaxCmin2]. -

Emtricitabine and Tenofovir Alafenamide
HIGHLIGHTS OF PRESCRIBING INFORMATION ------------------------DOSAGE AND ADMINISTRATION---------------------- These highlights do not include all the information needed to use • Testing: Prior to initiation of DESCOVY, patients should be tested for DESCOVY safely and effectively. See full prescribing information hepatitis B virus infection, and estimated creatinine clearance, urine for DESCOVY. glucose and urine protein should be obtained. (2.1) • ® Recommended dosage: One tablet taken once daily with or without DESCOVY (emtricitabine and tenofovir alafenamide) tablets, for food in patients with body weight at least 25 kg and a creatinine oral use clearance greater than or equal to 30 mL per minute. (2.2) Initial U.S. Approval: 2015 • Renal impairment: DESCOVY is not recommended in patients with WARNING: POST TREATMENT ACUTE EXACERBATION OF estimated creatinine clearance below 30 mL per minute. (2.3) HEPATITIS B ----------------------DOSAGE FORMS AND STRENGTHS-------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg of FTC and 25 mg of TAF (3) • DESCOVY is not approved for the treatment of chronic -------------------------------CONTRAINDICATIONS------------------------------ hepatitis B virus (HBV) infection. Severe acute None. exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have -----------------------WARNINGS AND PRECAUTIONS----------------------- discontinued products containing emtricitabine (FTC) • Immune reconstitution syndrome: May necessitate further evaluation and/or tenofovir disoproxil fumarate (TDF), and may occur and treatment.(5.2) with discontinuation of DESCOVY. Hepatic function should • New onset or worsening renal impairment: Assess creatinine be monitored closely in these patients. If appropriate, clearance, urine glucose, and urine protein in all patients before initiation of anti-hepatitis B therapy may be warranted. -

SELZENTRY (Maraviroc) Tablets, for Oral Use • Hepatotoxicity Accompanied by Severe Rash Or Systemic Allergic Reaction
---------------------------------- CONTRAINDICATIONS --------------------------------- HIGHLIGHTS OF PRESCRIBING INFORMATION • SELZENTRY is contraindicated in patients with severe renal impairment These highlights do not include all the information needed to use or end-stage renal disease (ESRD) (CrCl less than 30 mL per minute) SELZENTRY safely and effectively. See full prescribing information for who are concomitantly taking potent CYP3A inhibitors or inducers. (4) SELZENTRY. -------------------------- WARNINGS AND PRECAUTIONS -------------------------- SELZENTRY (maraviroc) tablets, for oral use • Hepatotoxicity accompanied by severe rash or systemic allergic reaction, SELZENTRY (maraviroc) oral solution including potentially life-threatening events, has been reported. Hepatic Initial U.S. Approval: 2007 laboratory parameters including ALT, AST, and bilirubin should be obtained prior to starting SELZENTRY and at other time points during WARNING: HEPATOTOXICITY See full prescribing information for complete boxed warning. treatment as clinically indicated. If rash or symptoms or signs of hepatitis or allergic reaction develop, hepatic laboratory parameters should be • Hepatotoxicity has been reported which may be preceded by severe monitored and discontinuation of treatment should be considered. When rash or other features of a systemic allergic reaction (e.g., fever, administering SELZENTRY to patients with pre-existing liver eosinophilia, or elevated IgE). (5.1) dysfunction or who are co-infected with hepatitis B and/or C virus, • -

SYMTUZA™ (Darunavir, Cobicistat, Emtricitabine, Tenofovir Alafenamide) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 9/20/2018 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: SYMTUZA™ (darunavir, cobicistat, emtricitabine, tenofovir alafenamide) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. -

Didanosine (Ddi) Trade Name Videx®, Videx EC® Class Nucleoside Reverse Transcriptase Inhibitor Molecular Weight 236.2
www.hiv-druginteractions.org Didanosine PK Fact Sheet Reviewed March 2016 Page 1 of 2 For personal use only. Not for distribution. For personal use only. Not for distribution. For personal use only. Not for distribution. Details Generic Name Didanosine (ddI) Trade Name Videx®, Videx EC® Class Nucleoside Reverse Transcriptase Inhibitor Molecular Weight 236.2 Structure O N HN N N HO CH2 O Summary of Key Pharmacokinetic Parameters Didanosine is enzymatically converted to dideoxyadenosine‐triphosphate (ddATP), its active metabolite. Linearity/non‐linearity Increases in plasma concentrations were dose proportional over the range of 50‐400 mg. Plasma half life ~1.4 h Cmax 933 ± 434 ng/ml (400 mg once daily, Videx EC) AUC 2432 ± 919 ng/ml.h (400 mg once daily, Videx EC) Bioavailability ~42% (following single dose of buffered formulation) Absorption To minimise the impact of food, Videx gastro‐resistant capsules should be administered on an empty stomach at least 2 hours before or 2 hours after a meal. Protein Binding <5% Volume of Distribution 54 L CSF:Plasma ratio ~21% of corresponding plasma concentration. Semen:Plasma ratio Median didanosine concentrations have been found to be greater in semen than in plasma: 455 ng/ml (range <50–2190 ng/ml) in seminal plasma and <50 (range <50–860 ng/ml) in blood. Large interindividual variability was noted [1]. Renal Clearance ~20% Renal Impairment Half‐life of didanosine after oral administration increased from an average of 1.4 hours in subjects with normal renal function to 4.1 hours in subjects with severe renal impairment requiring dialysis.